Wykres rodowodowy przedstawia dziedziczenie fenyloketonurii. Genetyczne choroby dziedziczne
Prawo rozszczepienia wyjaśnia również dziedziczenie fenyloketonurii (PKU), choroby, która rozwija się w wyniku nadmiaru ważnego aminokwasu fenyloalaniny (Phe) w organizmie człowieka. Nadmiar fenyloalaniny prowadzi do rozwoju upośledzenia umysłowego. Częstość występowania PKU jest stosunkowo niska (około 1 na 10 000 urodzeń), jednak na PKU cierpi około 1% osób z upośledzeniem umysłowym, co stanowi stosunkowo dużą grupę pacjentów, których upośledzenie umysłowe tłumaczy się jednorodnym mechanizmem genetycznym.
Podobnie jak w przypadku CG, badacze badali częstość występowania PKU w rodzinach probantów. Okazało się, że pacjenci chorzy na PKU mają zazwyczaj zdrowych rodziców. Ponadto zaobserwowano, że FKU występuje częściej w rodzinach, w których rodzice są spokrewnieni. Przykład rodziny probanda chorego na PKU przedstawiono na ryc. 2.3: chore dziecko urodziło się z fenotypowo zdrowych rodziców, którzy są spokrewnieni (kuzyni), ale siostra ojca dziecka choruje na PKU.
Ryż. 2.3. Przykład rodowodu rodziny, w której dziedziczona jest PKU (na tę chorobę choruje ciotka probanta).
Podwójna linia między małżonkami oznacza małżeństwo pokrewne.
Pozostałe symbole są takie same jak na ryc. 2.1.
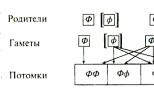
PKU jest przenoszona w sposób recesywny, tj. Genotyp pacjenta zawiera dwa allele PKU otrzymane od obojga rodziców. Potomkowie, którzy mają tylko jeden taki allel, nie chorują, ale są nosicielami allelu PKU” mogą przekazać chorobę swoim dzieciom. Na ryc. Rycina 2.4 przedstawia sposoby dziedziczenia alleli PKU od dwóch fenotypowo prawidłowych rodziców. Każdy rodzic ma jeden allel PKU i jeden allel prawidłowy. Prawdopodobieństwo, że każde dziecko odziedziczy allel PKU od każdego z rodziców, wynosi 50%. Prawdopodobieństwo, że dziecko odziedziczy allele PKU od obojga rodziców w tym samym czasie, wynosi 25\% (0,5 x 0,5 = 0,25; prawdopodobieństwa są mnożone, ponieważ zdarzenia dziedziczenia alleli od każdego z rodziców są od siebie niezależne).
Gen PKU i jego warianty strukturalne występujące w różnych populacjach zostały dobrze zbadane. Posiadana wiedza pozwala na terminową diagnostykę prenatalną w celu ustalenia, czy rozwijający się płód odziedziczył od obojga rodziców dwie kopie allelu PKU (fakt takiego dziedziczenia znacznie zwiększa prawdopodobieństwo wystąpienia choroby). W niektórych krajach, np. we Włoszech, gdzie zapadalność na PKU jest dość wysoka, taka diagnoza jest obowiązkowa dla każdej kobiety w ciąży.

Ryż. 2.4. Schemat krzyżowania: alleliczny mechanizm dziedziczenia PKU.
0 allel dominujący („zdrowy”); [f] allel recesywny powodujący rozwój choroby. FF, FF to dzieci fenotypowo normalne (75% z nich): tylko 25% ma prawidłowy genotyp (FF); kolejne 50\% jest fenotypowo zdrowych, ale jest nosicielami allelu PKU (Pf). Pozostałe 25% potomków jest chorych ([f][f])
Jak zauważono, PKU występuje częściej u osób zawierających związki małżeńskie z krewnymi. Chociaż częstość występowania PKU jest stosunkowo niska, około 1 na 50 osób jest nosicielami allelu PKU. Prawdopodobieństwo, że jeden nosiciel allelu PKU poślubi innego nosiciela takiego allelu, wynosi około 2%. Jednakże w przypadku zawarcia małżeństwa pomiędzy spokrewnionymi (tzn. jeśli małżonkowie należą do tego samego rodowodu, w którym dziedziczony jest allel PKU), prawdopodobieństwo, że oboje małżonkowie będą nosicielami allelu PKU i jednocześnie przekażą dwa allele nienarodzonemu dziecku, będzie stać się znacząco wyższe o 2\%.
Prawo rozszczepienia wyjaśnia również dziedziczenie fenyloketonurii
(PKU) – choroba, która rozwija się w wyniku nadmiaru ważnych
aminokwasy - fenyloalanina (Phe) w ludzkim ciele. Nadmiar
fenyloalanina prowadzi do rozwoju upośledzenia umysłowego. Częstotliwość
Częstość występowania PKU jest stosunkowo niska (około 1 na 10 000 nowych przypadków).
urodzonych), natomiast około 1% osób upośledzonych umysłowo
mov cierpi na PKU, co stanowi relatywnie więcej
największą grupę pacjentów, u których wyjaśniono upośledzenie umysłowe
jednorodny mechanizm genetyczny.
Podobnie jak w przypadku CG, badacze badali częstotliwość występowania
PKU w rodzinach probantów. Okazało się, że pacjenci cierpiący na PKU
zazwyczaj mają zdrowych rodziców. Poza tym zauważono, że
PKU występuje częściej w rodzinach, w których rodzice pochodzą z krwi
inni krewni. Przykład rodziny probanta chorego na PKU

Ryż. 2.3: chory
fenotypowy
zdrowy
rodzice-
krewni
cierpi
przekazywane
dziedzictwo,
chory
zawiera
otrzymane
rodzice.
Ryż. 2.3. Przykład rodowodu rodzinnego, w
cierpieć
przekazywane
choroba,
Czy
spadek (cierpi ciotka probanta
posiadacze alleli PKU i może
tę chorobę).
przekazać
Podwójna linia między małżonkami oznacza
Ryż. Pokazano 2.4
spokrewniony
Odpoczynek
tworzenie alleli PKU z dwóch
oznaczenia są takie same jak na rys. 2.1.
fenotypowo
normalna
rodzice.
leu ma jeden allel PKU i jeden allel normalny. Prawdopodobieństwo
że każde dziecko może odziedziczyć allel PKU od każdego
rodziców wynosi 50%. Prawdopodobieństwo, że dziecko jest
następuje po allelu PKU od obojga rodziców w tym samym czasie, wynosi 25%
(0,5 x 0,5 = 0,25; prawdopodobieństwa są mnożone w miarę dziedziczenia zdarzeń
allele od każdego z rodziców są od siebie niezależne).
Gen PKU i jego warianty strukturalne spotykane w różnych krajach
populacje zostały dobrze zbadane. Wiedza, którą dysponujemy jest
Ryż. 2.4. Schemat krzyżowania: alleliczny mechanizm dziedziczenia PKU.
F - allel dominujący („zdrowy”); [f] - powodujący allel recesywny
rozwój choroby. FF, FF – dzieci fenotypowo normalne (75% z nich); tylko
około 25% ma prawidłowy genotyp (FF); kolejne 50% jest fenotypowo zdrowych,
ale są nosicielami allelu PKU (FF). Pozostałe 25% potomków jest chorych
([f]).
małżeństwa, pozwalają na terminową diagnostykę prenatalną
tiki w celu ustalenia, czy rozwijający się zarodek odziedziczył
wdychać dwie kopie allelu PKU od obojga rodziców (fakt takiego dziedziczenia
vaniya gwałtownie zwiększa prawdopodobieństwo choroby). W niektórych krajach,
na przykład we Włoszech, gdzie częstość występowania PKU jest dość wysoka
sok, taka diagnostyka jest przeprowadzana bez wątpienia dla każdego
wydoić kobietę w ciąży.
Jak już wspomniano, PKU występuje częściej wśród osób wchodzących
zawiera związek małżeński z krewnymi. Pomimo tego, że spotkanie
Częstość występowania PKU jest stosunkowo niska i wynosi około 1 na 50 osób
nosiciel allelu PKU. Prawdopodobieństwo, że jeden nosiciel allelu
PKU poślubi innego nosiciela takiego allelu, tj
około 2%. Jednakże w przypadku zawarcia związku małżeńskiego między spokrewnionymi
krewni (tj. jeżeli małżonkowie należą do tego samego rodowodu, w
który allel PKU jest dziedziczony) prawdopodobieństwo, że
oboje małżonkowie będą nosicielami allelu PKU i jednocześnie nastąpi transfer
da nienarodzonemu dziecku dwa allele, będzie znacznie wyższy niż 2%.
 Genealogiczna metoda badania dziedziczności jest jedną z najstarszych i najczęściej stosowanych metod genetyki. Istotą metody jest sporządzenie rodowodów pozwalających na prześledzenie charakterystyki dziedziczenia cech. Metodę można zastosować, jeśli znani są bezpośredni krewni posiadacza badanej cechy w linii matczynej i ojcowskiej w kilku pokoleniach.
Genealogiczna metoda badania dziedziczności jest jedną z najstarszych i najczęściej stosowanych metod genetyki. Istotą metody jest sporządzenie rodowodów pozwalających na prześledzenie charakterystyki dziedziczenia cech. Metodę można zastosować, jeśli znani są bezpośredni krewni posiadacza badanej cechy w linii matczynej i ojcowskiej w kilku pokoleniach.

 Spis treści 1. 2. 3. 4. 5. Symbole Zasady sporządzania rodowodu Etapy rozwiązywania problemów Rodzaje dziedziczenia cech Rozwiązywanie problemów
Spis treści 1. 2. 3. 4. 5. Symbole Zasady sporządzania rodowodu Etapy rozwiązywania problemów Rodzaje dziedziczenia cech Rozwiązywanie problemów

 Zasady sporządzania rodowodów Osoba, od której rozpoczyna się sporządzanie rodowodu, nazywa się probantem. Bracia i siostry probanta nazywani są rodzeństwem. 1. Rodowód przedstawiono w taki sposób, aby każde pokolenie znajdowało się na własnej poziomej linii. Pokolenia są numerowane cyframi rzymskimi, a członkowie drzewa genealogicznego numerowani są cyframi arabskimi. 2. Sporządzanie rodowodu rozpoczyna się od probanda (w zależności od płci - kwadratu lub koła, oznaczonego strzałką), tak aby od niego można było wyprowadzić rodowód zarówno w dół, jak i w górę. 3. Obok probanda umieść symbole jego rodzeństwa w kolejności urodzenia (od lewej do prawej), łącząc je graficznym rockerem.
Zasady sporządzania rodowodów Osoba, od której rozpoczyna się sporządzanie rodowodu, nazywa się probantem. Bracia i siostry probanta nazywani są rodzeństwem. 1. Rodowód przedstawiono w taki sposób, aby każde pokolenie znajdowało się na własnej poziomej linii. Pokolenia są numerowane cyframi rzymskimi, a członkowie drzewa genealogicznego numerowani są cyframi arabskimi. 2. Sporządzanie rodowodu rozpoczyna się od probanda (w zależności od płci - kwadratu lub koła, oznaczonego strzałką), tak aby od niego można było wyprowadzić rodowód zarówno w dół, jak i w górę. 3. Obok probanda umieść symbole jego rodzeństwa w kolejności urodzenia (od lewej do prawej), łącząc je graficznym rockerem.
 4. Nad linią probanda wskaż rodziców, łącząc ich ze sobą linią małżeńską. 5. Na linii rodziców narysuj symbole najbliższych krewnych i ich małżonków, łącząc odpowiednio ich stopnie pokrewieństwa. 6. Na linii probanda wskaż jego kuzynów itp., braci i siostry, łącząc ich odpowiednio z linią rodziców. 7. Nad linią rodziców narysuj linię dziadków. 8. Jeżeli probant ma dzieci lub siostrzeńców, umieść ich w linii poniżej linii probanda.
4. Nad linią probanda wskaż rodziców, łącząc ich ze sobą linią małżeńską. 5. Na linii rodziców narysuj symbole najbliższych krewnych i ich małżonków, łącząc odpowiednio ich stopnie pokrewieństwa. 6. Na linii probanda wskaż jego kuzynów itp., braci i siostry, łącząc ich odpowiednio z linią rodziców. 7. Nad linią rodziców narysuj linię dziadków. 8. Jeżeli probant ma dzieci lub siostrzeńców, umieść ich w linii poniżej linii probanda.
 9. Po przedstawieniu rodowodu (lub jednocześnie z nim) należy odpowiednio wskazać właścicieli lub heterozygotycznych nosicieli cechy (najczęściej heterozygotycznych nosicieli określa się po zestawieniu i analizie rodowodu). 10. Wskaż (jeśli to możliwe) genotypy wszystkich członków rodowodu. 11. Jeżeli w rodzinie występuje kilka chorób dziedzicznych, które nie są ze sobą powiązane, utwórz rodowód dla każdej choroby osobno.
9. Po przedstawieniu rodowodu (lub jednocześnie z nim) należy odpowiednio wskazać właścicieli lub heterozygotycznych nosicieli cechy (najczęściej heterozygotycznych nosicieli określa się po zestawieniu i analizie rodowodu). 10. Wskaż (jeśli to możliwe) genotypy wszystkich członków rodowodu. 11. Jeżeli w rodzinie występuje kilka chorób dziedzicznych, które nie są ze sobą powiązane, utwórz rodowód dla każdej choroby osobno.
 Etapy rozwiązywania problemu 1. Określ rodzaj dziedziczenia cechy – dominujący lub recesywny. Aby to zrobić, dowiedz się: 1) czy badana cecha jest powszechna (we wszystkich pokoleniach, czy nie); 2) ilu członków rodu ma tę cechę; 3) czy zdarzają się przypadki urodzenia dzieci posiadających tę cechę, jeżeli rodzice tej cechy nie posiadają; 4) czy zdarzają się przypadki urodzenia dzieci bez badanej cechy, jeżeli posiadają ją oboje rodzice; 5) jaka część potomstwa nosi tę cechę w rodzinach, jeśli jeden z rodziców jest jej właścicielem.
Etapy rozwiązywania problemu 1. Określ rodzaj dziedziczenia cechy – dominujący lub recesywny. Aby to zrobić, dowiedz się: 1) czy badana cecha jest powszechna (we wszystkich pokoleniach, czy nie); 2) ilu członków rodu ma tę cechę; 3) czy zdarzają się przypadki urodzenia dzieci posiadających tę cechę, jeżeli rodzice tej cechy nie posiadają; 4) czy zdarzają się przypadki urodzenia dzieci bez badanej cechy, jeżeli posiadają ją oboje rodzice; 5) jaka część potomstwa nosi tę cechę w rodzinach, jeśli jeden z rodziców jest jej właścicielem.
 Etapy rozwiązywania problemu 2. Ustal, czy cecha jest dziedziczona w sposób sprzężony z płcią. Aby to zrobić, dowiedz się: 1) jak często objaw występuje u osób obu płci; jeśli jest to rzadkie, to która płeć występuje częściej; 2) osoby danej płci dziedziczą cechę od ojca i matki noszących tę cechę.
Etapy rozwiązywania problemu 2. Ustal, czy cecha jest dziedziczona w sposób sprzężony z płcią. Aby to zrobić, dowiedz się: 1) jak często objaw występuje u osób obu płci; jeśli jest to rzadkie, to która płeć występuje częściej; 2) osoby danej płci dziedziczą cechę od ojca i matki noszących tę cechę.
 Etapy rozwiązywania problemu 3. Na podstawie wyników analizy spróbuj określić genotypy wszystkich członków rodowodu. Aby określić genotypy, przede wszystkim znajdź wzór na podział potomków w jednym pokoleniu.
Etapy rozwiązywania problemu 3. Na podstawie wyników analizy spróbuj określić genotypy wszystkich członków rodowodu. Aby określić genotypy, przede wszystkim znajdź wzór na podział potomków w jednym pokoleniu.
 Rodzaje dziedziczenia cechy. 1. Dziedziczenie autosomalne dominujące: 1) cecha występuje często w rodowodzie, niemal we wszystkich pokoleniach, równie często u chłopców i dziewcząt; 2) jeśli jedno z rodziców jest nosicielem cechy, to cecha ta pojawi się albo u całego potomstwa, albo u połowy.
Rodzaje dziedziczenia cechy. 1. Dziedziczenie autosomalne dominujące: 1) cecha występuje często w rodowodzie, niemal we wszystkich pokoleniach, równie często u chłopców i dziewcząt; 2) jeśli jedno z rodziców jest nosicielem cechy, to cecha ta pojawi się albo u całego potomstwa, albo u połowy.
 Jaskra jest chorobą oczu charakteryzującą się podwyższonym ciśnieniem wewnątrzgałkowym i zmniejszoną ostrością wzroku. Czynnikami ryzyka rozwoju jaskry są: dziedziczność, cukrzyca, miażdżyca, urazy oczu, choroby zapalne i zwyrodnieniowe oczu. Przy stale podwyższonym ciśnieniu wewnątrzgałkowym stopniowo rozwija się zanik nerwu wzrokowego, a osoba traci wzrok. Brachydaktylia (brachydaktylia; brachy- + grecki palec daktylos; synonim krótki palec) to anomalia rozwojowa: skrócenie palców u rąk i nóg. dziedziczona w sposób autosomalny dominujący.
Jaskra jest chorobą oczu charakteryzującą się podwyższonym ciśnieniem wewnątrzgałkowym i zmniejszoną ostrością wzroku. Czynnikami ryzyka rozwoju jaskry są: dziedziczność, cukrzyca, miażdżyca, urazy oczu, choroby zapalne i zwyrodnieniowe oczu. Przy stale podwyższonym ciśnieniu wewnątrzgałkowym stopniowo rozwija się zanik nerwu wzrokowego, a osoba traci wzrok. Brachydaktylia (brachydaktylia; brachy- + grecki palec daktylos; synonim krótki palec) to anomalia rozwojowa: skrócenie palców u rąk i nóg. dziedziczona w sposób autosomalny dominujący.
 Rodzaje dziedziczenia cechy. 2. Dziedziczenie autosomalne recesywne: 1) cecha jest rzadka, nie we wszystkich pokoleniach, jednakowo częsta zarówno u chłopców, jak i dziewcząt; 2) cecha może ujawnić się u dzieci, nawet jeśli rodzice tej cechy nie posiadają; 3) jeśli jedno z rodziców jest nosicielem cechy, to nie pojawi się ona u dzieci lub pojawi się u połowy potomstwa.
Rodzaje dziedziczenia cechy. 2. Dziedziczenie autosomalne recesywne: 1) cecha jest rzadka, nie we wszystkich pokoleniach, jednakowo częsta zarówno u chłopców, jak i dziewcząt; 2) cecha może ujawnić się u dzieci, nawet jeśli rodzice tej cechy nie posiadają; 3) jeśli jedno z rodziców jest nosicielem cechy, to nie pojawi się ona u dzieci lub pojawi się u połowy potomstwa.
 Co to jest fenyloketonuria? Fenyloketonuria (PKU) to choroba dziedziczna, która zwiększa ilość aminokwasu fenyloalaniny we krwi do szkodliwego poziomu. (Aminokwasy są budulcem białek). Jeśli PKU nie jest leczone, nadmiar fenyloalaniny może powodować upośledzenie umysłowe i inne poważne problemy zdrowotne. W jaki sposób ludzie dziedziczą PKU? PKU dziedziczy się w sposób autosomalny recesywny, co oznacza, że aby dana osoba mogła zachorować, należy zmienić dwie kopie genu. Najczęściej rodzice dziecka z chorobą autosomalną recesywną nie są dotknięci chorobą, ale są nosicielami jednej kopii zmienionego genu.
Co to jest fenyloketonuria? Fenyloketonuria (PKU) to choroba dziedziczna, która zwiększa ilość aminokwasu fenyloalaniny we krwi do szkodliwego poziomu. (Aminokwasy są budulcem białek). Jeśli PKU nie jest leczone, nadmiar fenyloalaniny może powodować upośledzenie umysłowe i inne poważne problemy zdrowotne. W jaki sposób ludzie dziedziczą PKU? PKU dziedziczy się w sposób autosomalny recesywny, co oznacza, że aby dana osoba mogła zachorować, należy zmienić dwie kopie genu. Najczęściej rodzice dziecka z chorobą autosomalną recesywną nie są dotknięci chorobą, ale są nosicielami jednej kopii zmienionego genu.
 Rodzaje dziedziczenia cechy. 3. Dziedziczenie związane z płcią: 1) X - dziedziczenie dominujące: ü cecha występuje częściej u kobiet; ü jeśli matka jest chora, a ojciec zdrowy, wówczas cecha przekazywana jest potomstwu niezależnie od płci, może objawiać się zarówno u dziewcząt, jak i chłopców; ü jeśli matka jest zdrowa, a ojciec chory, objawy wystąpią u wszystkich córek, ale u synów nie.
Rodzaje dziedziczenia cechy. 3. Dziedziczenie związane z płcią: 1) X - dziedziczenie dominujące: ü cecha występuje częściej u kobiet; ü jeśli matka jest chora, a ojciec zdrowy, wówczas cecha przekazywana jest potomstwu niezależnie od płci, może objawiać się zarówno u dziewcząt, jak i chłopców; ü jeśli matka jest zdrowa, a ojciec chory, objawy wystąpią u wszystkich córek, ale u synów nie.
 3. Dziedziczenie związane z płcią: 2) X - dziedziczenie recesywne: cecha częściej występuje u mężczyzn; Częściej objaw objawia się po pokoleniu; Jeśli oboje rodzice są zdrowi, ale matka jest heterozygotą, wówczas cecha ta często pojawia się u 50% synów; Jeśli ojciec jest chory, a matka jest heterozygotą, to kobiety również mogą mieć tę cechę.
3. Dziedziczenie związane z płcią: 2) X - dziedziczenie recesywne: cecha częściej występuje u mężczyzn; Częściej objaw objawia się po pokoleniu; Jeśli oboje rodzice są zdrowi, ale matka jest heterozygotą, wówczas cecha ta często pojawia się u 50% synów; Jeśli ojciec jest chory, a matka jest heterozygotą, to kobiety również mogą mieć tę cechę.

 3. Dziedziczenie związane z płcią: 3) Dziedziczenie związane z Y: ücecha występuje tylko u mężczyzn; Jeśli ojciec ma jakąś cechę, to z reguły wszyscy synowie również ją posiadają.
3. Dziedziczenie związane z płcią: 3) Dziedziczenie związane z Y: ücecha występuje tylko u mężczyzn; Jeśli ojciec ma jakąś cechę, to z reguły wszyscy synowie również ją posiadają.
 Przykład rozwiązania problemu Probanda jest kobietą praworęczną. Jej dwie siostry są praworęczne, a dwaj bracia leworęczni. Matka jest praworęczna. Ma dwóch braci i siostrę, wszyscy praworęczni. Babcia i dziadek są praworęczni. Ojciec probanda jest leworęczny, jego siostra i brat są leworęczni, pozostali dwaj bracia i siostra są praworęczni. Rozwiązanie: 1. Narysuj symbol probanda. Pokazujemy obecność znaku w probancie.
Przykład rozwiązania problemu Probanda jest kobietą praworęczną. Jej dwie siostry są praworęczne, a dwaj bracia leworęczni. Matka jest praworęczna. Ma dwóch braci i siostrę, wszyscy praworęczni. Babcia i dziadek są praworęczni. Ojciec probanda jest leworęczny, jego siostra i brat są leworęczni, pozostali dwaj bracia i siostra są praworęczni. Rozwiązanie: 1. Narysuj symbol probanda. Pokazujemy obecność znaku w probancie.
 2. Symbole jej rodzeństwa umieszczamy obok symbolu probanda. Łączymy je graficznym rockerem.
2. Symbole jej rodzeństwa umieszczamy obok symbolu probanda. Łączymy je graficznym rockerem.




 7. Określ genotypy członków rodowodu. Znak praworęczności pojawia się w każdym pokoleniu zarówno u kobiet, jak i u mężczyzn. Wskazuje to na autosomalny dominujący typ dziedziczenia cechy. I A- A- II A- A- A- Aa aa A- III aa Aa Aa A- aa
7. Określ genotypy członków rodowodu. Znak praworęczności pojawia się w każdym pokoleniu zarówno u kobiet, jak i u mężczyzn. Wskazuje to na autosomalny dominujący typ dziedziczenia cechy. I A- A- II A- A- A- Aa aa A- III aa Aa Aa A- aa
 Zadanie 2. Na podstawie rodowodu pokazanego na rysunku określ charakter przejawu cechy oznaczonej kolorem czarnym (dominująca, recesywna, związana z płcią lub nie). Określ genotyp rodziców i dzieci w pierwszym pokoleniu.
Zadanie 2. Na podstawie rodowodu pokazanego na rysunku określ charakter przejawu cechy oznaczonej kolorem czarnym (dominująca, recesywna, związana z płcią lub nie). Określ genotyp rodziców i dzieci w pierwszym pokoleniu.
 Schemat rozwiązania problemu: 1) Cecha recesywna nie jest powiązana z płcią; 2) Genotypy rodziców: matka - aa, ojciec - AA lub Aa 3) Genotypy dzieci: heterozygotyczny syn i córka - Aa.
Schemat rozwiązania problemu: 1) Cecha recesywna nie jest powiązana z płcią; 2) Genotypy rodziców: matka - aa, ojciec - AA lub Aa 3) Genotypy dzieci: heterozygotyczny syn i córka - Aa.
 Zadanie 3 Korzystając z rodowodu pokazanego na diagramie, ustal rodzaj i charakter przejawów cechy zaznaczonej na czarno (dominująca, recesywna, związana z płcią lub niezwiązana). Określ genotypy dzieci w pierwszym pokoleniu.
Zadanie 3 Korzystając z rodowodu pokazanego na diagramie, ustal rodzaj i charakter przejawów cechy zaznaczonej na czarno (dominująca, recesywna, związana z płcią lub niezwiązana). Określ genotypy dzieci w pierwszym pokoleniu.
 Schemat rozwiązania problemu: 1) Cecha jest recesywna, powiązana z chromosomem X; 2) Genotypy rodziców: matka – XHA, ojciec – XAU; 3) Genotypy dzieci w F 1: syn – Ha. Ech, córka - HAHA córka - HAHA
Schemat rozwiązania problemu: 1) Cecha jest recesywna, powiązana z chromosomem X; 2) Genotypy rodziców: matka – XHA, ojciec – XAU; 3) Genotypy dzieci w F 1: syn – Ha. Ech, córka - HAHA córka - HAHA
 Zadanie 4 Korzystając z rodowodu osoby pokazanego na rysunku, ustal charakter dziedziczenia cechy „małe oczy”, zaznaczonej na czarno (dominująca lub recesywna, sprzężona z płcią lub nie). Określ genotypy rodziców i potomstwa F 1 (1, 2, 3, 4, 5). 1 2 3 4 5
Zadanie 4 Korzystając z rodowodu osoby pokazanego na rysunku, ustal charakter dziedziczenia cechy „małe oczy”, zaznaczonej na czarno (dominująca lub recesywna, sprzężona z płcią lub nie). Określ genotypy rodziców i potomstwa F 1 (1, 2, 3, 4, 5). 1 2 3 4 5
 Schemat rozwiązania problemu: 1) Cecha jest recesywna, a nie związana z płcią; 2) Genotypy rodziców: matka – Aa, ojciec – Aa; 3) Genotypy potomków w F 1: 1, 2 – Aa, 3, 5 – AA lub Aa; 4 – aa.
Schemat rozwiązania problemu: 1) Cecha jest recesywna, a nie związana z płcią; 2) Genotypy rodziców: matka – Aa, ojciec – Aa; 3) Genotypy potomków w F 1: 1, 2 – Aa, 3, 5 – AA lub Aa; 4 – aa.
 Kodyfikator elementów treści w biologii 3.4 Genetyka, jej zadania. Dziedziczność i zmienność są właściwościami organizmów. Metody genetyczne. Podstawowe pojęcia i symbolika genetyczna. Chromosomalna teoria dziedziczności. Współczesne poglądy na temat genu i genomu. 3. 5 Wzorce dziedziczności, ich podstawy cytologiczne. Wzorce dziedziczenia ustalone przez G. Mendla, ich podstawy cytologiczne (krzyżowanie mono- i dihybrydowe). Prawa Morgana: sprzężone dziedziczenie cech, zaburzenie sprzężenia genowego. Genetyka seksu. Dziedziczenie cech sprzężonych z płcią. Interakcja genów. Genotyp jako system integralny. Genetyka człowieka. Metody badania genetyki człowieka. Rozwiązywanie problemów genetycznych. Sporządzanie schematów przejazdów.
Kodyfikator elementów treści w biologii 3.4 Genetyka, jej zadania. Dziedziczność i zmienność są właściwościami organizmów. Metody genetyczne. Podstawowe pojęcia i symbolika genetyczna. Chromosomalna teoria dziedziczności. Współczesne poglądy na temat genu i genomu. 3. 5 Wzorce dziedziczności, ich podstawy cytologiczne. Wzorce dziedziczenia ustalone przez G. Mendla, ich podstawy cytologiczne (krzyżowanie mono- i dihybrydowe). Prawa Morgana: sprzężone dziedziczenie cech, zaburzenie sprzężenia genowego. Genetyka seksu. Dziedziczenie cech sprzężonych z płcią. Interakcja genów. Genotyp jako system integralny. Genetyka człowieka. Metody badania genetyki człowieka. Rozwiązywanie problemów genetycznych. Sporządzanie schematów przejazdów.
 SPECYFIKACJA pracy egzaminacyjnej z biologii A 7. Genetyka, jej zadania, podstawowe pojęcia genetyczne. A 8. Wzorce dziedziczności. Genetyka człowieka. A 9. Wzorce zmienności. A 30. Wzorce genetyczne. Wpływ mutagenów na aparat genetyczny komórek i organizmów. C 6. Rozwiązywanie problemów z genetyki w celu zastosowania wiedzy w nowej sytuacji.
SPECYFIKACJA pracy egzaminacyjnej z biologii A 7. Genetyka, jej zadania, podstawowe pojęcia genetyczne. A 8. Wzorce dziedziczności. Genetyka człowieka. A 9. Wzorce zmienności. A 30. Wzorce genetyczne. Wpływ mutagenów na aparat genetyczny komórek i organizmów. C 6. Rozwiązywanie problemów z genetyki w celu zastosowania wiedzy w nowej sytuacji.
 Część A 1. Genetyka ma ogromne znaczenie dla medycyny, ponieważ 1) zwalcza epidemie 2) tworzy leki do leczenia pacjentów 3) ustala przyczyny chorób dziedzicznych 4) chroni środowisko przed zanieczyszczeniem mutagenami
Część A 1. Genetyka ma ogromne znaczenie dla medycyny, ponieważ 1) zwalcza epidemie 2) tworzy leki do leczenia pacjentów 3) ustala przyczyny chorób dziedzicznych 4) chroni środowisko przed zanieczyszczeniem mutagenami
 2. Metoda stosowana do badania charakteru przejawów cech u sióstr lub braci, które rozwinęły się z jednego zapłodnionego jaja, nazywa się 1. 2. 3. 4. Hybrydologiczny genealogiczny bliźniak cytogenetyczny
2. Metoda stosowana do badania charakteru przejawów cech u sióstr lub braci, które rozwinęły się z jednego zapłodnionego jaja, nazywa się 1. 2. 3. 4. Hybrydologiczny genealogiczny bliźniak cytogenetyczny
 3. Metodę genealogiczną wykorzystuje się do 1) uzyskiwania mutacji genowych i genomowych 2) badania wpływu wychowania na ontogenezę człowieka 3) badania dziedzicznych chorób człowieka 4) badania etapów ewolucji świata organicznego
3. Metodę genealogiczną wykorzystuje się do 1) uzyskiwania mutacji genowych i genomowych 2) badania wpływu wychowania na ontogenezę człowieka 3) badania dziedzicznych chorób człowieka 4) badania etapów ewolucji świata organicznego
 4. Jaka jest funkcja medycznych konsultacji genetycznych dla par rodziców? 1. Identyfikuje predyspozycje rodziców do chorób zakaźnych 2. Określa możliwość posiadania bliźniąt 3. Określa prawdopodobieństwo wystąpienia chorób dziedzicznych u dzieci 4. Identyfikuje predyspozycje rodziców do zaburzeń metabolicznych
4. Jaka jest funkcja medycznych konsultacji genetycznych dla par rodziców? 1. Identyfikuje predyspozycje rodziców do chorób zakaźnych 2. Określa możliwość posiadania bliźniąt 3. Określa prawdopodobieństwo wystąpienia chorób dziedzicznych u dzieci 4. Identyfikuje predyspozycje rodziców do zaburzeń metabolicznych
 Określ genotyp według fenotypu Kolor oczu u osoby jest określany przez gen autosomalny; ślepota barw jest genem recesywnym powiązanym z płcią. Określ genotyp kobiety brązowookiej, normalnie widzącej kolory, której ojciec jest daltonistą (brązowe oczy dominują nad niebieskookimi) 1) AAXDXD 3) Aa. Xd 2) Aaa. XDXd 4) aa. XDXd
Określ genotyp według fenotypu Kolor oczu u osoby jest określany przez gen autosomalny; ślepota barw jest genem recesywnym powiązanym z płcią. Określ genotyp kobiety brązowookiej, normalnie widzącej kolory, której ojciec jest daltonistą (brązowe oczy dominują nad niebieskookimi) 1) AAXDXD 3) Aa. Xd 2) Aaa. XDXd 4) aa. XDXd
 Część C Rozwiązywanie problemów genetycznych związanych z zastosowaniem wiedzy w nowej sytuacji: krzyżowanie dihybrydowe, dziedziczenie cech sprzężonych z płcią, dziedziczenie sprzężone cech (z krzyżowaniem, bez krzyżowania), oznaczanie grup krwi, analiza rodowodu
Część C Rozwiązywanie problemów genetycznych związanych z zastosowaniem wiedzy w nowej sytuacji: krzyżowanie dihybrydowe, dziedziczenie cech sprzężonych z płcią, dziedziczenie sprzężone cech (z krzyżowaniem, bez krzyżowania), oznaczanie grup krwi, analiza rodowodu
 Część C U ludzi dziedziczenie albinizmu nie jest powiązane z płcią (A – obecność melaniny w komórkach skóry i – brak melaniny w komórkach skóry – albinizm), a hemofilia jest sprzężona z płcią (XH – prawidłowe krzepnięcie krwi , Xh – hemofilia). Określ genotypy rodziców oraz możliwe genotypy, płeć i fenotypy dzieci ze związku kobiety dihomozygotycznej, prawidłowej pod względem obu alleli, oraz mężczyzny-albinosa chorego na hemofilię. Zrób diagram rozwiązania problemu.
Część C U ludzi dziedziczenie albinizmu nie jest powiązane z płcią (A – obecność melaniny w komórkach skóry i – brak melaniny w komórkach skóry – albinizm), a hemofilia jest sprzężona z płcią (XH – prawidłowe krzepnięcie krwi , Xh – hemofilia). Określ genotypy rodziców oraz możliwe genotypy, płeć i fenotypy dzieci ze związku kobiety dihomozygotycznej, prawidłowej pod względem obu alleli, oraz mężczyzny-albinosa chorego na hemofilię. Zrób diagram rozwiązania problemu.
 Schemat rozwiązania problemu obejmuje: 1) genotypy rodziców: ♀AAXHXH (gamety AXH); ♂aa. Xh. Y (gamety a. Xh, a. Y); 2) genotypy i płeć dzieci: ♀Aa. XHXh; ♂Aa. XHY; 3) fenotypy dzieci: dziewczynka, która na zewnątrz ma oba allele, ale jest nosicielką genów powodujących albinizm i hemofilię; Chłopiec, który na zewnątrz ma oba allele w normie, ale jest nosicielem genu albinizmu.
Schemat rozwiązania problemu obejmuje: 1) genotypy rodziców: ♀AAXHXH (gamety AXH); ♂aa. Xh. Y (gamety a. Xh, a. Y); 2) genotypy i płeć dzieci: ♀Aa. XHXh; ♂Aa. XHY; 3) fenotypy dzieci: dziewczynka, która na zewnątrz ma oba allele, ale jest nosicielką genów powodujących albinizm i hemofilię; Chłopiec, który na zewnątrz ma oba allele w normie, ale jest nosicielem genu albinizmu.
Anomalie prowadzące do podwyższonych poziomów fenyloalanina krew, najczęściej niedobór hydroksylazy fenyloalaniny (PAH) lub fenyloketonurię (PKU), ilustrują niemal wszystkie zasady genetyki biochemicznej związane z defektami enzymatycznymi. Wszystkie nieprawidłowości genetyczne metabolizmu fenyloalaniny są wynikiem mutacji powodujących utratę funkcji w genie kodującym PAH lub w genach niezbędnych do syntezy lub odtworzenia jego kofaktora, BH4.
Klasyczna fenyloketonuria(PKU) słusznie uważa się za wzorowego przedstawiciela wrodzonych wad metabolizmu. Jest to autosomalne recesywne zaburzenie rozkładu fenyloalaniny spowodowane mutacjami w genie kodującym PAH – enzym przekształcający fenyloalaninę w tyrozynę. Odkrycie fenyloketonurii (PKU) przez Fehlinga w 1934 r. jako pierwsze wykazało, że przyczyną upośledzenia umysłowego jest defekt genetyczny.
Z powodu braku możliwości recyklingu fenyloalanina pacjenci z fenyloketonurią (PKU) kumulują ten aminokwas w płynach ustrojowych. Hiperfenyloalaninemia uszkadza rozwijający się ośrodkowy układ nerwowy we wczesnym dzieciństwie i zaburza funkcjonowanie dojrzałego mózgu. Niewielka część fenyloalaniny jest metabolizowana alternatywnymi szlakami, w wyniku czego powstają zwiększone ilości kwasu fenylopirogronowego (kwasu ketonowego, od którego pochodzi nazwa choroby) i innych metabolitów wydalanych z moczem.
To ciekawe, że chociaż defekt enzymatyczny jest znany od dziesięcioleci, dokładny mechanizm patogenetyczny tego, jak zwiększona fenyloalanina uszkadza mózg, jest nadal nieznany. Co ważne, rozwojowi uszkodzeń neurologicznych spowodowanych blokiem metabolicznym w klasycznej PKU można w dużym stopniu zapobiec poprzez zmiany diety zapobiegające kumulacji fenyloalaniny. Leczenie fenyloketonurii (PKU) stało się modelowym sposobem leczenia wielu chorób metabolicznych, których przebieg można poprawić poprzez zapobieganie kumulacji substratu enzymu i jego pochodnych.
Badania przesiewowe noworodków w kierunku fenyloketonurii (PKU)
Populacja jest szeroko stosowana ekranizacja noworodków na fenyloketonurię (PKU). Fenyloketonuria (PKU) jest przykładem chorób genetycznych, w przypadku których uzasadnione są masowe badania przesiewowe noworodków; choroba ta występuje stosunkowo często w wielu populacjach (do 1 na 2900 żywych noworodków). Leczenie rozpoczęte na wczesnym etapie życia jest bardzo skuteczne; bez leczenia nieuchronnie rozwija się ciężkie upośledzenie umysłowe. Badania przesiewowe wykonuje się kilka dni po urodzeniu.
Kropla krwi uzyskana z nakłucia obcasy, nałożono na bibułę filtracyjną, wysuszono i przesłano do scentralizowanego laboratorium w celu oceny poziomu fenyloalaniny we krwi i stosunku fenyloalaniny do tyrozyny. W przeszłości próbki pobierano przed wypisem dziecka ze szpitala. Trend w kierunku wcześniejszego wypisania matki i noworodka po porodzie zmienił tę praktykę. Zaleca się nie wykonywać badania przed 24. godziną życia, ponieważ poziom fenyloalaniny w fenyloketonurii (PKU) wzrasta dopiero po urodzeniu. Pozytywne wyniki badań należy szybko potwierdzić, ponieważ opóźnienie rozpoczęcia leczenia o więcej niż 4 tygodnie po porodzie nie pozwala uniknąć wpływu na stan intelektualny pacjentek chorych na fenyloketonurię (PKU).
Różne formy fenyloketonurii i hiperfenyloalaninemii
Ponieważ (PKU) wiąże się z poważnym niedoborem aktywności hydroksylazy fenyloalaniny (PAH) (mniej niż 1% w porównaniu z grupą kontrolną), zmutowany PAH z resztkową aktywnością powoduje mniej poważne objawy fenotypowe, tak zwaną hiperfenyloalaninemię i atypową fenyloketonurię (PKU).
Hiperfenyloalaninemia fenyloketonurię (PKU), inną niż fenyloketonurię (PKU), rozpoznaje się, jeśli stężenie fenyloalaniny w osoczu wynosi poniżej 1 mmol/l przy normalnej diecie. Ten stopień hiperfenyloalaninemii jest tylko 10 razy wyższy niż normalnie i znacznie niższy niż stężenia stwierdzane w klasycznej fenyloketonurii (PKU) (> 1 mmol/l). Umiarkowany wzrost fenyloalaniny w hiperfenyloalaninemii prawdopodobnie nie zaszkodzi funkcjonowaniu mózgu, a nawet może być korzystny, jeśli wzrost jest niewielki (<0,4 ммоль), такие дети обращают на себя внимание врачей только благодаря скринингу. Их нормальный фенотип оказался наилучшим показателем безопасного уровня фенилаланина плазмы, который не следует превышать при лечении пациентов с классической фенилкетонурии (ФКУ).
Nietypowy(PKU) – kategoria obejmująca pacjentów ze stężeniem fenyloalaniny pośrednim pomiędzy klasyczną PKU a hiperfenyloalaninemią; tacy pacjenci wymagają pewnego ograniczenia fenyloalaniny w diecie, ale w mniejszym stopniu niż u pacjentów z klasyczną fenyloketonurią (PKU). Przykładem heterogeniczności klinicznej jest kompleks tych trzech fenotypów klinicznych z mutacjami w genie PAH.
Hiperfenyloalaninemia: heterogeniczność alleli i locus w fenyloketonurii (PKU)
Molekularny wady w genie hydroksylazy fenyloalaniny. Pacjenci z hiperfenyloalaninemią, w tym klasyczną fenyloketonurią (PKU), atypową fenyloketonurią (PKU) i łagodną hiperfenyloalaninemią, wykazują uderzający stopień heterogeniczności alleli w locus hydroksylazy fenyloalaniny (PAH) (ponad 400 różnych mutacji na całym świecie).
Zdecydowana większość alleli hydroksylaza fenyloalaniny(PAH) to dość rzadkie mutacje, które zakłócają właściwości enzymatyczne hydroksylazy fenyloalaniny (PAH) i prowadzą do hiperfenyloalaninemii, chociaż stwierdzono również łagodne polimorfizmy lub mniej powszechne łagodne warianty.
W populacjach Europejskie pochodzenie około dwie trzecie znanych zmutowanych chromosomów jest reprezentowanych przez sześć mutacji. Sześć innych mutacji jest odpowiedzialnych za nieco ponad 80% mutacji hydroksylazy fenyloalaniny (PAH) w populacjach azjatyckich. Inne mutacje patogenne są mniej powszechne. Aby informacje te były powszechnie dostępne, międzynarodowe konsorcjum opracowało bazę danych dotyczącą mutacji w genie hydroksylazy fenyloalaniny (PAH).
We wszystkim populacje Występuje wyraźna heterogeniczność genetyczna hydroksylazy fenyloalaniny (PAH). Ze względu na wysoki stopień heterogeniczności alleli w miejscu, większość pacjentów z fenyloketonurią (PKU) w wielu populacjach to heterozygoty złożone (tj. mają dwa różne allele patogenne), co jest w pełni zgodne z obserwowaną heterogenicznością enzymatyczną i fenotypową u zaburzenia hydroksylazy fenyloalaniny (PAH).

Na początku wydawało się, że to znajomość genotypu hydroksylaza fenyloalaniny(FA) wiarygodnie przewiduje szczegóły fenotypu; oczekiwanie to nie było w pełni uzasadnione, choć stwierdzono pewną korelację pomiędzy genotypem WWA a fenotypem biochemicznym.
Ogólnie rzecz biorąc, mutacje, które całkowicie tłumią lub dramatycznie zmniejszają aktywność hydroksylaza fenyloalaniny(PAH) powodują klasyczną fenyloketonurię (PKU), natomiast mutacje powodujące wystarczająco dużą resztkową aktywność enzymatyczną są powiązane z łagodnymi fenotypami.
Jednak pewne mutacje hydroksylaza fenyloalaniny(FA) u pacjentów homozygotycznych określa całe spektrum fenotypów, od klasycznej fenyloketonurii (PKU) po łagodną hiperfenyloalaninemię.
Zatem stało się oczywiste, że w formacji fenotyp obserwowane w konkretnym genotypie, w grę wchodzą inne niezidentyfikowane czynniki biologiczne, do których niewątpliwie zaliczają się geny modyfikujące. Obserwacja ta, obecnie uznawana za wspólną cechę wielu chorób jednogenowych, wskazuje, że nawet choroby jednogenowe, takie jak fenyloketonuria (PKU), nie są chorobami prostymi genetycznie.
Wady metabolizmu tetrahydrobiopteryny w fenyloketonurii (PKU)
Początkowo uważano, że wszystkie dzieci są dziedziczne hiperfenyloalaninemia u pacjenta występuje pierwotny niedobór hydroksylazy fenyloalaniny (PAH). Obecnie jest jasne, że około 1–3% pacjentów ma prawidłowy gen PAH, a ich hiperfenyloalaninemia jest wynikiem defektu genetycznego jednego z kilku innych genów zaangażowanych w syntezę lub regenerację kofaktora PAH, BH4. Związek jednego fenotypu, takiego jak hiperfenyloalaninemia, z mutacjami w różnych genach jest przykładem heterogeniczności locus.
Jak pokazują mutacje w genach kodujących białka hydroksylaza fenyloalaniny(PAH) i metabolizm jej kofaktora, biopteryny, białek kodowanych przez geny wykazujące heterogeniczność locus, zwykle uczestniczą w tym samym łańcuchu reakcji biochemicznych. Po raz pierwszy zidentyfikowano pacjentów z niedoborem BH4, ponieważ pomimo skutecznego utrzymywania niskiego stężenia fenyloalaniny w diecie, wcześnie rozwinęły się u nich poważne problemy neurologiczne.
Słabe wyniki zostały częściowo wyjaśnione zapotrzebowanie na kofaktor BH4 na aktywność dwóch innych enzymów, hydroksylazy tyrozynowej i hydroksylazy tryptofanu. Obie te hydroksylazy odgrywają kluczową rolę w syntezie neuroprzekaźników monoaminowych, takich jak dehydroksyfenyloalanina, norepinefryna, epinefryna i serotonina. Pacjenci z niedoborem BH4 mają upośledzoną biosyntezę BH4 lub regenerację BH4. Podobnie jak klasyczna fenyloketonuria (PKU), choroba ta jest dziedziczona w sposób autosomalny recesywny.
Ważne jest, aby odróżnić pacjentów z zaburzeniami metabolizmu BH4 od pacjentów z mutacjami hydroksylaza fenyloalaniny(FA), ponieważ ich leczenie znacznie się różni. Po pierwsze, ponieważ struktura białkowa hydroksylazy fenyloalaniny (PAH) jest prawidłowa u pacjentów z zaburzeniami BH4, jej aktywność może zostać przywrócona, jeśli pacjenci ci otrzymają duże dawki BH4, co prowadzi do obniżenia poziomu fenyloalaniny w osoczu. Dzięki temu stopień ograniczenia fenyloalaniny w diecie pacjentów z zaburzeniami metabolizmu BH4 może zostać znacząco zmniejszony, a część pacjentów może przejść na dietę normalną (tj. bez ograniczenia fenyloalaniny).
Po drugie, trzeba też spróbować normalizować poziomu neuroprzekaźników w mózgu tych pacjentów poprzez podawanie produktów hydroksylazy tyrozynowej i hydroksylazy tryptofanu: odpowiednio L-dopa i 5-hydroksytryptofan. Z tych powodów wszystkie noworodki z hiperfenyloalaninemią należy badać pod kątem nieprawidłowości w metabolizmie BH4.
Reakcja na tetrahydrobiopterynę z mutacjami w genie PAH w fenyloketonurii (PKU)
U większości pacjentów z mutacjami w genie hydroksylaza fenyloalaniny(PAH), a nie w metabolizmie BH4, podczas doustnego podawania dużych dawek kofaktora hydroksylazy fenyloalaniny (PAH) BH4 zaobserwowano wyraźne obniżenie poziomu fenyloalaniny we krwi. Pacjenci ze znaczną resztkową aktywnością hydroksylazy fenyloalaniny (PAH) (tj. pacjenci z atypową fenyloketonurią (PKU) i hiperfenyloalaninemią) najlepiej reagują na takie leczenie, ale niewielka liczba pacjentów, nawet z klasyczną fenyloketonurią (PKU), również reaguje na to leczenie. Jednocześnie obecność resztkowej aktywności WWA nie gwarantuje wpływu na stężenie fenyloalaniny w osoczu, gdy przepisywany jest BH4.
Najprawdopodobniej chodzi o stopień reakcji reakcje na BH4 zależy od specyficznych właściwości każdego zmutowanego białka hydroksylazy fenyloalaniny (PAH), odzwierciedlając leżącą u podstaw heterogeniczności allelicznej mutacji PAH. Wykazano, że wprowadzenie BH4 do diety ma działanie terapeutyczne poprzez kilka mechanizmów spowodowanych zwiększeniem ilości prawidłowego kofaktora wchodzącego w kontakt ze zmutowanym.
Mechanizmy te obejmują stabilizację mutanta enzym, ochrona enzymu przed degradacją komórek, zwiększone dostarczanie kofaktora do enzymu, który ma niskie powinowactwo do BH4 i inne korzystne skutki w zakresie właściwości kinetycznych i katalitycznych enzymu. Dostarczanie zwiększonej ilości kofaktora jest powszechną strategią stosowaną w leczeniu wielu wrodzonych wad metabolizmu.
Szereg mutacji genowych, w których zmieniana jest struktura tylko jednego genu, prowadzi do rozwoju upośledzenia umysłowego. Według niektórych szacunków u 7–10% pacjentów z upośledzeniem umysłowym jest ono spowodowane tego typu mutacjami.
Zespół reakcji biochemicznych zachodzących w organizmie nazywa się metabolizmem. Wiele genów koduje białka, które jako enzymy uczestniczą w niektórych reakcjach metabolicznych. Mutacja w takim genie może doprowadzić do tego, że organizm zacznie wytwarzać mniej aktywny lub całkowicie nieaktywny enzym, a czasami do całkowitego zaprzestania syntezy enzymu. W tym przypadku reakcja zwykle przeprowadzana przez ten enzym albo zwalnia, albo w ogóle nie zachodzi, co powoduje odpowiednie zaburzenie dziedziczne - jeden z tak zwanych wrodzonych błędów metabolizmu. Do najczęstszych chorób dziedzicznych o podłożu genetycznym zalicza się fenyloketonurię, anemię sierpowatokrwinkową, chorobę Tay-Sachsa, hemofilię i cukrzycę. Stopień, w jakim wpływają one na fenotyp, zależy od tego, jak ważny jest dany enzym dla organizmu. Widzieliśmy powyżej, że choroba Tay-Sachsa i mukowiscydoza prowadzą do śmierci. Niektóre inne nieprawidłowości genetyczne powodują różne poważne problemy w organizmie, ale nie są śmiertelne.
Fenyloketonuria i albinizm wpływają na ten sam szlak metaboliczny.
Fenyloketonuria to choroba, w której w wyniku mutacji zostaje zaburzona struktura enzymu biorącego udział w metabolizmie aminokwasu fenyloalaniny (hydroksylazy fenyloalaniny). Enzym ten jest niezbędny do konwersji fenyloalaniny do tyrozyny. Choroby tego rodzaju nazywane są enzymopatiami, tj. spowodowane defektem enzymów. W przypadku tej choroby fenyloalanina i produkty jej nieprawidłowego metabolizmu (kwas fenylooctowy) gromadzą się we krwi, co prowadzi do uszkodzenia rozwijającego się układu nerwowego. Jest to głównie zniszczenie mieliny i zwyrodnienie gąbczastego układu nerwowego. Występuje upośledzenie umysłowe, małogłowie, psychoza, drżenie, drgawki i spastyczność.
Fenyloketonuria dotyka osoby homozygotyczne pod względem genu recesywnego, który pozbawia je zdolności do syntezy jednego z enzymów niezbędnych do przekształcenia aminokwasu fenyloalaniny w inny aminokwas – tyrozynę. Zamiast przekształcać się w tyrozynę, fenyloalanina przekształca się w kwas fenylopirogronowy, który gromadzi się w toksycznych ilościach we krwi, wpływa na mózg i (jeśli nie jest szybko leczony) powoduje upośledzenie umysłowe. Mocz pacjentów zawiera również kwas fenylopirogronowy, który nadaje mu charakterystyczny zapach. Obecnie fenyloketonurię leczy się specjalną dietą. Aby to zrobić, w pierwszych latach życia dziecka fenyloalanina jest prawie całkowicie wykluczona z jego diety. Po zakończeniu rozwoju mózgu pacjentkę chorą na fenyloketonurię rozpoczyna się na normalnej diecie, natomiast kobieta z tym zaburzeniem genetycznym powinna podczas ciąży stosować dietę ubogą w fenyloalaninę, aby zapobiec nieprawidłowemu rozwojowi mózgu płodu. W wielu stanach Stanów Zjednoczonych wszystkie noworodki muszą przejść specjalne badania w kierunku PKU i innych wrodzonych wad metabolizmu.
Osobnikom homozygotycznym pod względem genu albinizmu brakuje enzymu, który normalnie katalizuje konwersję tyrozyny do melaniny, tj. pigment określający brązową lub czarną barwę oczu, włosów i skóry. Albinosy mają białe włosy oraz bardzo jasną skórę i oczy. Naturalnie może pojawić się pytanie, czy pacjenci chorzy na fenyloketonurię są także albinosami, skoro ich organizm nie wytwarza tyrozyny, z której ostatecznie powstaje melanina. Jednak tacy pacjenci nie są albinosami, ponieważ tyrozyna nie tylko powstaje w samym organizmie z fenyloalaniny, ale także dostaje się do organizmu z pożywieniem. To prawda, że pacjenci z fenyloketonurią mają zwykle jasne oczy, jasną karnację i jasne włosy. Oczywiście mogą być wśród nich albinosy, ale tylko wtedy, gdy osobnik jest homozygotą pod względem obu genów recesywnych.
Dziedziczenie fenyloketonurii (PKU) wyjaśnia prawo podziału. Mutacja ta jest recesywna, tj. może stawić opór fenotypowi tylko w stanie homozygotycznym. Największą zapadalność na fenyloketonurię odnotowano w Irlandii (16,4 przypadków na 100 tys. noworodków); dla porównania: w USA – 5 przypadków na 100 tys. noworodków.
Gen PKU i jego warianty strukturalne występujące w różnych populacjach zostały dobrze zbadane. Posiadana wiedza pozwala na terminową diagnostykę prenatalną w celu ustalenia, czy rozwijający się zarodek odziedziczył od obojga rodziców dwie kopie allelu PKU (fakt takiego dziedziczenia znacznie zwiększa prawdopodobieństwo wystąpienia choroby). W niektórych krajach, np. we Włoszech, gdzie zapadalność na PKU jest dość wysoka, taka diagnoza jest obowiązkowa dla każdej kobiety w ciąży.
PKU występuje częściej u osób zawierających związki małżeńskie z krewnymi. Chociaż częstość występowania PKU jest stosunkowo niska, około 1 na 50 osób jest nosicielami allelu PKU. Prawdopodobieństwo, że jeden nosiciel allelu PKU poślubi innego nosiciela takiego allelu, wynosi około 2%. Jeżeli jednak małżeństwo następuje pomiędzy spokrewnionymi krewnymi (tj. jeżeli małżonkowie należą do tego samego rodowodu, w którym dziedziczony jest allel PKU), prawdopodobieństwo, że oboje małżonkowie będą nosicielami allelu PKU i jednocześnie przekażą w przyszłości dwa allele dziecka będzie znacznie wyższa o 2%.
W przypadku fenyloketonurii mamy uderzający przykład tego, jak można zapobiec rozwojowi choroby o charakterze genetycznym, wybierając czynniki środowiskowe. Obecnie fenyloketonurię można łatwo wykryć podczas rutynowych badań noworodków w 2-3 dniu życia (normalnie stężenie fenyloalaniny w osoczu krwi nie powinno przekraczać 4 mg/dl). Pacjentom przepisuje się dietę ubogą w fenyloalaninę, co pozwala uniknąć rozwojowych uszkodzeń układu nerwowego. W tym przypadku tyrozyna staje się aminokwasem niezbędnym i należy zadbać o jej obecność w diecie. Najbardziej krytycznym okresem są wczesne etapy ontogenezy, dlatego w wieku dorosłym wiele osób nie przestrzega już ograniczeń dietetycznych, chociaż jest to nadal pożądane. Kobiety chore na fenyloketonurię, niezależnie od swojej choroby, muszą w czasie ciąży przestrzegać specjalnej diety, w przeciwnym razie wysoki poziom fenyloalaniny we krwi będzie miał szkodliwy wpływ na rozwijający się płód.
Fenyloketonuria jest dobrym przykładem interakcji genotyp-środowisko. Istotą tej choroby jest różna wrażliwość osobników o różnych genotypach na wpływy środowiska. To samo środowisko (w tym przypadku środowisko to charakter odżywiania) powoduje u niektórych genotypów ciężką chorobę (fenyloketonurię), podczas gdy u innych genotypów nie obserwuje się absolutnie żadnych zmian patologicznych. W innych warunkach środowiskowych (pod warunkiem stosowania specjalnej diety) zanikają różnice między genotypami w zakresie tej cechy (fenyloketonuria).