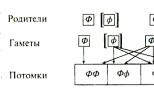
แผนภูมิสายเลือดแสดงการถ่ายทอดทางพันธุกรรมของฟีนิลคีโตนูเรีย โรคทางพันธุกรรมทางพันธุกรรม
กฎแห่งความแตกแยกยังอธิบายถึงการถ่ายทอดทางพันธุกรรมของฟีนิลคีโตนูเรีย (PKU) ซึ่งเป็นโรคที่พัฒนาขึ้นอันเป็นผลมาจากกรดอะมิโนฟีนิลอะลานีน (เพ) ที่มากเกินไปในร่างกายมนุษย์ ฟีนิลอะลานีนที่มากเกินไปทำให้เกิดภาวะปัญญาอ่อน อุบัติการณ์ของ PKU ค่อนข้างต่ำ (ประมาณ 1 ใน 10,000 การเกิด) อย่างไรก็ตาม ประมาณ 1% ของบุคคลที่มีความบกพร่องทางสติปัญญาต้องทนทุกข์ทรมานจาก PKU ดังนั้นจึงเป็นกลุ่มผู้ป่วยที่ค่อนข้างใหญ่ซึ่งมีการอธิบายภาวะปัญญาอ่อนโดยกลไกทางพันธุกรรมที่เป็นเนื้อเดียวกัน
เช่นเดียวกับในกรณีของ CG นักวิจัยได้ศึกษาอุบัติการณ์ของ PKU ในครอบครัวของกลุ่มโพรแบนด์ ปรากฎว่าผู้ป่วยที่เป็นโรค PKU มักจะมีพ่อแม่ที่มีสุขภาพดี นอกจากนี้ยังพบว่า FKU พบได้บ่อยในครอบครัวที่พ่อแม่เป็นญาติทางสายเลือด ตัวอย่างของครอบครัว proband ที่ทุกข์ทรมานจาก PKU แสดงไว้ในรูปที่ 1 2.3: เด็กป่วยเกิดจากพ่อแม่ที่มีสุขภาพแข็งแรงดีซึ่งเป็นญาติทางสายเลือด (ลูกพี่ลูกน้อง) แต่น้องสาวของพ่อของเด็กป่วยด้วย PKU
ข้าว. 2.3. ตัวอย่างสายเลือดของครอบครัวที่สืบทอด PKU (ป้าของ proband ป่วยด้วยโรคนี้)
เส้นคู่ระหว่างคู่สมรสหมายถึงการแต่งงานในสายเลือดเดียวกัน
สัญลักษณ์ที่เหลือจะเหมือนกับในรูป 2.1.
PKU ถูกส่งโดยโหมดการสืบทอดแบบถอยเช่น จีโนไทป์ของผู้ป่วยประกอบด้วยอัลลีล PKU สองตัวที่ได้รับจากพ่อแม่ทั้งสอง ลูกหลานที่มีอัลลีลดังกล่าวเพียงอันเดียวจะไม่เป็นโรคนี้ แต่เป็นพาหะของอัลลีล PKU" สามารถส่งต่อไปยังลูกหลานได้ ในรูป รูปที่ 2.4 แสดงวิธีการสืบทอดอัลลีล PKU จากพ่อแม่ที่มีฟีโนไทป์ปกติสองคน ผู้ปกครองแต่ละคนมีอัลลีล PKU หนึ่งตัวและอัลลีลปกติหนึ่งตัว ความน่าจะเป็นที่เด็กแต่ละคนจะได้รับอัลลีล PKU จากผู้ปกครองแต่ละคนคือ 50% ความน่าจะเป็นที่เด็กจะได้รับอัลลีล PKU จากทั้งพ่อและแม่ในเวลาเดียวกันคือ 25\% (0.5 x 0.5 = 0.25 ความน่าจะเป็นจะถูกคูณเนื่องจากเหตุการณ์ในการสืบทอดอัลลีลจากพ่อและแม่แต่ละคนมีความเป็นอิสระจากกัน)
ยีน PKU และโครงสร้างที่แตกต่างกันซึ่งพบในประชากรที่แตกต่างกันได้รับการศึกษาอย่างดี ความรู้ที่มีอยู่ช่วยให้เราสามารถวินิจฉัยก่อนคลอดได้ทันท่วงทีเพื่อตรวจสอบว่าทารกในครรภ์ที่กำลังพัฒนาได้รับอัลลีล PKU สองสำเนาจากพ่อแม่ทั้งสองหรือไม่ (ความจริงของการถ่ายทอดทางพันธุกรรมดังกล่าวเพิ่มโอกาสในการเกิดโรคอย่างรวดเร็ว) ในบางประเทศ เช่น อิตาลี ซึ่งมีอุบัติการณ์ของ PKU ค่อนข้างสูง การวินิจฉัยดังกล่าวเป็นสิ่งจำเป็นสำหรับหญิงตั้งครรภ์ทุกคน

ข้าว. 2.4. รูปแบบการข้าม: กลไกอัลลีลิกของการสืบทอด PKU
0 อัลลีลที่โดดเด่น (“ สุขภาพดี”); [f] อัลลีลถอยที่ทำให้เกิดการพัฒนาของโรค FF, FF เป็นเด็กปกติทางฟีโนไทป์ (75% ในจำนวนนี้): มีเพียง 25% เท่านั้นที่มีจีโนไทป์ปกติ (FF); อีก 50\% มีฟีโนไทป์ที่ดีต่อสุขภาพ แต่เป็นพาหะของอัลลีล PKU (Pf) ลูกหลานที่เหลืออีก 25% ป่วย ([f][f])
ตามที่ระบุไว้ PKU พบได้บ่อยกว่าในกลุ่มผู้ที่แต่งงานกับญาติทางสายเลือด แม้ว่าอุบัติการณ์ของ PKU จะค่อนข้างต่ำ แต่ประมาณ 1 ใน 50 คนเป็นพาหะของ PKU allele ความน่าจะเป็นที่พาหะของอัลลีล PKU จะแต่งงานกับพาหะของอัลลีลอีกตัวหนึ่งคือประมาณ 2\% อย่างไรก็ตาม เมื่อแต่งงานระหว่างญาติทางสายเลือด (เช่น หากคู่สมรสอยู่ในสายเลือดเดียวกันซึ่งได้รับอัลลีล PKU) ความเป็นไปได้ที่คู่สมรสทั้งสองจะเป็นพาหะของอัลลีล PKU และส่งต่ออัลลีลสองตัวไปยังเด็กในครรภ์พร้อมกัน สูงขึ้นอย่างมีนัยสำคัญ 2\ %
กฎแห่งความแตกแยกยังอธิบายการถ่ายทอดทางพันธุกรรมของฟีนิลคีโตนูเรียด้วย
(PKU) - โรคที่เกิดจากสิ่งที่สำคัญมากเกินไป
กรดอะมิโน - ฟีนิลอะลานีน (เพ)ในร่างกายมนุษย์ ส่วนเกิน
ฟีนิลอะลานีนนำไปสู่การพัฒนาของภาวะปัญญาอ่อน ความถี่
อุบัติการณ์ของ PKU ค่อนข้างต่ำ (ประมาณ 1 ใน 10,000 ใหม่
เกิด) อย่างไรก็ตาม ประมาณ 1% ของบุคคลที่มีความบกพร่องทางสติปัญญา
mov ต้องทนทุกข์ทรมานจาก PKU จึงทำให้ค่อนข้างมากขึ้น
กลุ่มผู้ป่วยที่ใหญ่ที่สุดที่มีการอธิบายภาวะปัญญาอ่อน
กลไกทางพันธุกรรมที่เป็นเนื้อเดียวกัน
เช่นเดียวกับในกรณีของ CG นักวิจัยได้ศึกษาความถี่ของการเกิดขึ้น
PKU ในตระกูลโพรแบนด์ ปรากฎว่าผู้ป่วยเป็นโรค PKU
มักจะมีพ่อแม่ที่มีสุขภาพดี นอกจากนี้ก็สังเกตเห็นว่า
PKU พบได้บ่อยในครอบครัวที่พ่อแม่มีสายเลือด
ญาติคนอื่น ๆ. ตัวอย่างครอบครัว Proband ที่ป่วยเป็นโรค PKU

ข้าว. 2.3: ป่วย
ฟีโนไทป์
สุขภาพดี
ผู้ปกครอง-
ญาติ
ทนทุกข์ทรมาน
ส่ง
มรดก
ป่วย
ประกอบด้วย
ได้รับ
ผู้ปกครอง.
ข้าว. 2.3.ตัวอย่างสายเลือดครอบครัวใน
ทุกข์ทรมาน
ส่ง
การเจ็บป่วย,
เป็น
มรดก (ป้าของโพรแบนด์ทนทุกข์ทรมาน
ผู้ถืออัลลีล PKU และกระป๋อง
โรคนี้)
ส่งมอบ
เส้นคู่ระหว่างคู่สมรสหมายถึง
ข้าว. 2.4 แสดง
อยู่ในตระกูลเดียวกัน
พักผ่อน
การก่อตัวของอัลลีล PKU จากสอง
การกำหนดจะเหมือนกับในรูป 2.1.
ฟีโนไทป์
ปกติ
ผู้ปกครอง.
leu มีอัลลีล PKU หนึ่งอันและอัลลีลปกติหนึ่งอัน ความน่าจะเป็น
ที่เด็กทุกคนสามารถสืบทอดอัลลีล PKU จากทุกคนได้
ของผู้ปกครองคือ 50% ความน่าจะเป็นที่เด็กจะเป็น
ติดตามอัลลีล PKU จากทั้งพ่อและแม่พร้อมกันคือ 25%
(0.5 x 0.5 = 0.25 ความน่าจะเป็นจะคูณตามเหตุการณ์ที่สืบทอดมา
อัลลีลจากพ่อแม่แต่ละคนจะเป็นอิสระจากกัน)
ยีน PKU และโครงสร้างที่แตกต่างกันพบว่ามีความแตกต่างกัน
ประชากรได้รับการศึกษาเป็นอย่างดี ความรู้ที่เราจำหน่ายก็คือ
ข้าว. 2.4.รูปแบบการข้าม: กลไกอัลลีลิกของการสืบทอด PKU
F - อัลลีลที่โดดเด่น (“ สุขภาพดี”); [f] - ก่อให้เกิดอัลลีลแบบถอย
การพัฒนาของโรค FF, FF - เด็กปกติทางฟีโนไทป์ (75% ของพวกเขา); เท่านั้น
ประมาณ 25% มีจีโนไทป์ปกติ (FF); อีก 50% มีฟีโนไทป์ที่ดีต่อสุขภาพ
แต่เป็นพาหะของอัลลีล PKU (FF) ลูกหลานที่เหลืออีก 25% ป่วย
([ฉ][ฉ])
การแต่งงานเพื่อให้สามารถวินิจฉัยก่อนคลอดได้ทันท่วงที
สำบัดสำนวนเพื่อตรวจสอบว่าตัวอ่อนที่กำลังพัฒนาได้รับการสืบทอดมาหรือไม่
หายใจเอาอัลลีล PKU สองชุดจากทั้งพ่อและแม่ (ข้อเท็จจริงของมรดกดังกล่าว
วานิยาเพิ่มโอกาสเป็นโรคได้อย่างรวดเร็ว) ในบางประเทศ
เช่นในประเทศอิตาลีซึ่งมีอุบัติการณ์ของ PKU ค่อนข้างสูง
น้ำผลไม้การวินิจฉัยดังกล่าวจะดำเนินการโดยไม่ล้มเหลวในแต่ละครั้ง
รีดนมหญิงตั้งครรภ์
ตามที่ระบุไว้แล้ว PKU นั้นพบได้บ่อยในหมู่ผู้ที่เข้ามา
แต่งงานกับญาติทางสายเลือด ทั้งๆ ที่การประชุมนั้น
อุบัติการณ์ของ PKU ค่อนข้างต่ำ ประมาณ 1 ใน 50 คนคือ
พาหะของอัลลีล PKU ความน่าจะเป็นที่พาหะของอัลลีลหนึ่งราย
PKU จะแต่งงานกับผู้ให้บริการรายอื่นของอัลลีลดังกล่าว
ประมาณ 2% แต่เมื่อแต่งงานกันระหว่างคนในตระกูลเดียวกัน
ญาติ (เช่น หากคู่สมรสอยู่ในสายเลือดเดียวกัน ใน
อัลลีล PKU ใดที่สืบทอดมา) ความน่าจะเป็นนั้น
คู่สมรสทั้งสองจะเป็นพาหะของอัลลีล PKU และถ่ายโอนพร้อมกัน
จะให้อัลลีลสองตัวแก่เด็กในครรภ์ซึ่งจะสูงกว่า 2% อย่างมีนัยสำคัญ
 วิธีการลำดับวงศ์ตระกูลในการศึกษาเกี่ยวกับพันธุกรรมเป็นหนึ่งในวิธีการทางพันธุศาสตร์ที่เก่าแก่และใช้กันอย่างแพร่หลายมากที่สุด สาระสำคัญของวิธีการนี้คือการรวบรวมสายเลือดที่ช่วยให้สามารถติดตามลักษณะของการสืบทอดลักษณะต่างๆ ได้ วิธีการนี้ใช้ได้หากทราบญาติสายตรงของเจ้าของลักษณะที่ศึกษาในสายมารดาและบิดาในหลายชั่วอายุคน
วิธีการลำดับวงศ์ตระกูลในการศึกษาเกี่ยวกับพันธุกรรมเป็นหนึ่งในวิธีการทางพันธุศาสตร์ที่เก่าแก่และใช้กันอย่างแพร่หลายมากที่สุด สาระสำคัญของวิธีการนี้คือการรวบรวมสายเลือดที่ช่วยให้สามารถติดตามลักษณะของการสืบทอดลักษณะต่างๆ ได้ วิธีการนี้ใช้ได้หากทราบญาติสายตรงของเจ้าของลักษณะที่ศึกษาในสายมารดาและบิดาในหลายชั่วอายุคน

 สารบัญ 1. 2. 3. 4. 5. สัญลักษณ์ กฎสำหรับการวาดสายเลือด ขั้นตอนการแก้ปัญหา ประเภทของการสืบทอดลักษณะ การแก้ปัญหา
สารบัญ 1. 2. 3. 4. 5. สัญลักษณ์ กฎสำหรับการวาดสายเลือด ขั้นตอนการแก้ปัญหา ประเภทของการสืบทอดลักษณะ การแก้ปัญหา

 กฎเกณฑ์ในการรวบรวมสายเลือด บุคคลที่เริ่มรวบรวมสายเลือดเรียกว่าโพรแบนด์ พี่น้องของโพรแบนด์เรียกว่าพี่น้อง 1. มีการแสดงสายเลือดเพื่อให้แต่ละรุ่นมีเส้นแนวนอนของตัวเอง รุ่นต่างๆ จะถูกกำหนดหมายเลขด้วยเลขโรมัน และสมาชิกของลำดับวงศ์ตระกูลจะถูกกำหนดหมายเลขด้วยเลขอารบิค 2. การวาดสายเลือดเริ่มต้นจาก proband (ขึ้นอยู่กับเพศ - สี่เหลี่ยมหรือวงกลมระบุด้วยลูกศร) เพื่อที่จะสามารถวาดสายเลือดจากเขาได้ทั้งขึ้นและลง 3. ถัดจากโพรแบนด์ ให้วางสัญลักษณ์ของพี่น้องตามลำดับเกิด (จากซ้ายไปขวา) โดยเชื่อมเข้ากับกราฟิคร็อคเกอร์
กฎเกณฑ์ในการรวบรวมสายเลือด บุคคลที่เริ่มรวบรวมสายเลือดเรียกว่าโพรแบนด์ พี่น้องของโพรแบนด์เรียกว่าพี่น้อง 1. มีการแสดงสายเลือดเพื่อให้แต่ละรุ่นมีเส้นแนวนอนของตัวเอง รุ่นต่างๆ จะถูกกำหนดหมายเลขด้วยเลขโรมัน และสมาชิกของลำดับวงศ์ตระกูลจะถูกกำหนดหมายเลขด้วยเลขอารบิค 2. การวาดสายเลือดเริ่มต้นจาก proband (ขึ้นอยู่กับเพศ - สี่เหลี่ยมหรือวงกลมระบุด้วยลูกศร) เพื่อที่จะสามารถวาดสายเลือดจากเขาได้ทั้งขึ้นและลง 3. ถัดจากโพรแบนด์ ให้วางสัญลักษณ์ของพี่น้องตามลำดับเกิด (จากซ้ายไปขวา) โดยเชื่อมเข้ากับกราฟิคร็อคเกอร์
 4. เหนือเส้นโปรแบนด์ ระบุผู้ปกครอง เชื่อมต่อกันด้วยเส้นสมรส 5. บนเส้นผู้ปกครอง ให้วาดสัญลักษณ์ของญาติสนิทและคู่สมรสของพวกเขา เพื่อเชื่อมโยงระดับความสัมพันธ์ของพวกเขาตามลำดับ 6. ในบรรทัดของ proband ให้ระบุลูกพี่ลูกน้อง ฯลฯ พี่น้อง และเชื่อมโยงพวกเขาตามสายของผู้ปกครอง 7. เหนือเส้นพ่อแม่ ให้ลากเส้นปู่ย่าตายาย 8. หาก proband มีลูกหรือหลานชาย ให้วางไว้บนบรรทัดใต้เส้นของ proband
4. เหนือเส้นโปรแบนด์ ระบุผู้ปกครอง เชื่อมต่อกันด้วยเส้นสมรส 5. บนเส้นผู้ปกครอง ให้วาดสัญลักษณ์ของญาติสนิทและคู่สมรสของพวกเขา เพื่อเชื่อมโยงระดับความสัมพันธ์ของพวกเขาตามลำดับ 6. ในบรรทัดของ proband ให้ระบุลูกพี่ลูกน้อง ฯลฯ พี่น้อง และเชื่อมโยงพวกเขาตามสายของผู้ปกครอง 7. เหนือเส้นพ่อแม่ ให้ลากเส้นปู่ย่าตายาย 8. หาก proband มีลูกหรือหลานชาย ให้วางไว้บนบรรทัดใต้เส้นของ proband
 9. หลังจากแสดงภาพสายเลือด (หรือพร้อมกันกับสายเลือด) ให้แสดงเจ้าของหรือพาหะแบบเฮเทอโรไซกัสของลักษณะอย่างเหมาะสม (ส่วนใหญ่มักจะระบุพาหะแบบเฮเทอโรไซกัสหลังจากการรวบรวมและวิเคราะห์สายเลือด) 10. ระบุ (ถ้าเป็นไปได้) จีโนไทป์ของสมาชิกทุกคนในสายเลือด 11. หากมีโรคทางพันธุกรรมหลายอย่างในครอบครัวที่ไม่เกี่ยวข้องกัน ให้สร้างสายเลือดสำหรับแต่ละโรคแยกกัน
9. หลังจากแสดงภาพสายเลือด (หรือพร้อมกันกับสายเลือด) ให้แสดงเจ้าของหรือพาหะแบบเฮเทอโรไซกัสของลักษณะอย่างเหมาะสม (ส่วนใหญ่มักจะระบุพาหะแบบเฮเทอโรไซกัสหลังจากการรวบรวมและวิเคราะห์สายเลือด) 10. ระบุ (ถ้าเป็นไปได้) จีโนไทป์ของสมาชิกทุกคนในสายเลือด 11. หากมีโรคทางพันธุกรรมหลายอย่างในครอบครัวที่ไม่เกี่ยวข้องกัน ให้สร้างสายเลือดสำหรับแต่ละโรคแยกกัน
 ขั้นตอนของการแก้ปัญหา 1. กำหนดประเภทของการสืบทอดลักษณะ - เด่นหรือถอย เมื่อต้องการทำเช่นนี้ ให้ค้นหา: 1) ลักษณะที่กำลังศึกษาอยู่เป็นเรื่องธรรมดาหรือไม่ (ในทุกรุ่นหรือไม่); 2) มีสมาชิกสายเลือดกี่คนที่มีลักษณะ 3) มีกรณีการเกิดของเด็กที่มีลักษณะนี้หรือไม่หากผู้ปกครองไม่แสดงลักษณะนี้ 4) มีกรณีการเกิดของเด็กที่ไม่มีลักษณะการศึกษาหรือไม่หากมีทั้งพ่อและแม่ 5) ส่วนใดของลูกหลานที่มีลักษณะในครอบครัวถ้าพ่อแม่คนใดคนหนึ่งเป็นเจ้าของ
ขั้นตอนของการแก้ปัญหา 1. กำหนดประเภทของการสืบทอดลักษณะ - เด่นหรือถอย เมื่อต้องการทำเช่นนี้ ให้ค้นหา: 1) ลักษณะที่กำลังศึกษาอยู่เป็นเรื่องธรรมดาหรือไม่ (ในทุกรุ่นหรือไม่); 2) มีสมาชิกสายเลือดกี่คนที่มีลักษณะ 3) มีกรณีการเกิดของเด็กที่มีลักษณะนี้หรือไม่หากผู้ปกครองไม่แสดงลักษณะนี้ 4) มีกรณีการเกิดของเด็กที่ไม่มีลักษณะการศึกษาหรือไม่หากมีทั้งพ่อและแม่ 5) ส่วนใดของลูกหลานที่มีลักษณะในครอบครัวถ้าพ่อแม่คนใดคนหนึ่งเป็นเจ้าของ
 ขั้นตอนของการแก้ปัญหา 2. พิจารณาว่าลักษณะดังกล่าวได้รับการถ่ายทอดมาในลักษณะที่เชื่อมโยงกับเพศหรือไม่ ในการทำเช่นนี้ค้นหา: 1) อาการนี้เกิดขึ้นในคนทั้งสองเพศบ่อยแค่ไหน; ถ้ามันหายากแล้วเพศไหนที่ถือมันบ่อยกว่า 2) บุคคลที่เพศสืบทอดลักษณะมาจากบิดาและมารดาผู้มีลักษณะดังกล่าว
ขั้นตอนของการแก้ปัญหา 2. พิจารณาว่าลักษณะดังกล่าวได้รับการถ่ายทอดมาในลักษณะที่เชื่อมโยงกับเพศหรือไม่ ในการทำเช่นนี้ค้นหา: 1) อาการนี้เกิดขึ้นในคนทั้งสองเพศบ่อยแค่ไหน; ถ้ามันหายากแล้วเพศไหนที่ถือมันบ่อยกว่า 2) บุคคลที่เพศสืบทอดลักษณะมาจากบิดาและมารดาผู้มีลักษณะดังกล่าว
 ขั้นตอนของการแก้ปัญหา 3. พยายามกำหนดจีโนไทป์ของสมาชิกสายเลือดทั้งหมดจากผลการวิเคราะห์ ในการกำหนดจีโนไทป์ ก่อนอื่นให้ค้นหาสูตรสำหรับการแยกผู้สืบทอดในรุ่นเดียว
ขั้นตอนของการแก้ปัญหา 3. พยายามกำหนดจีโนไทป์ของสมาชิกสายเลือดทั้งหมดจากผลการวิเคราะห์ ในการกำหนดจีโนไทป์ ก่อนอื่นให้ค้นหาสูตรสำหรับการแยกผู้สืบทอดในรุ่นเดียว
 ประเภทของการสืบทอดลักษณะ 1. Autosomal dominant inheritance: 1) ลักษณะนี้เกิดขึ้นบ่อยครั้งในสายเลือด ในเกือบทุกรุ่น บ่อยเท่ากันในทั้งเด็กชายและเด็กหญิง; 2) หากผู้ปกครองคนใดคนหนึ่งเป็นพาหะของลักษณะ ลักษณะนี้จะปรากฏในลูกหลานทั้งหมดหรือครึ่งหนึ่ง
ประเภทของการสืบทอดลักษณะ 1. Autosomal dominant inheritance: 1) ลักษณะนี้เกิดขึ้นบ่อยครั้งในสายเลือด ในเกือบทุกรุ่น บ่อยเท่ากันในทั้งเด็กชายและเด็กหญิง; 2) หากผู้ปกครองคนใดคนหนึ่งเป็นพาหะของลักษณะ ลักษณะนี้จะปรากฏในลูกหลานทั้งหมดหรือครึ่งหนึ่ง
 โรคต้อหินเป็นโรคทางตาที่มีลักษณะความดันลูกตาเพิ่มขึ้นและการมองเห็นลดลง ปัจจัยเสี่ยงในการเกิดโรคต้อหิน ได้แก่ กรรมพันธุ์ เบาหวาน หลอดเลือด การบาดเจ็บที่ตา โรคตาอักเสบและเสื่อม เมื่อความดันในลูกตาสูงขึ้นอย่างต่อเนื่อง การฝ่อของเส้นประสาทตาจะค่อยๆ พัฒนาขึ้น และบุคคลนั้นสูญเสียการมองเห็น Brachydactyly (brachydactylia; brachy- + Greek daktylos finger; synonym short-fingered) เป็นความผิดปกติของพัฒนาการ: นิ้วหรือนิ้วเท้าสั้นลง สืบทอดมาในลักษณะเด่นแบบออโตโซม
โรคต้อหินเป็นโรคทางตาที่มีลักษณะความดันลูกตาเพิ่มขึ้นและการมองเห็นลดลง ปัจจัยเสี่ยงในการเกิดโรคต้อหิน ได้แก่ กรรมพันธุ์ เบาหวาน หลอดเลือด การบาดเจ็บที่ตา โรคตาอักเสบและเสื่อม เมื่อความดันในลูกตาสูงขึ้นอย่างต่อเนื่อง การฝ่อของเส้นประสาทตาจะค่อยๆ พัฒนาขึ้น และบุคคลนั้นสูญเสียการมองเห็น Brachydactyly (brachydactylia; brachy- + Greek daktylos finger; synonym short-fingered) เป็นความผิดปกติของพัฒนาการ: นิ้วหรือนิ้วเท้าสั้นลง สืบทอดมาในลักษณะเด่นแบบออโตโซม
 ประเภทของการสืบทอดลักษณะ 2. การถ่ายทอดทางพันธุกรรมแบบถอยอัตโนมัติ (Autosomal recessive inheritance) 1) ลักษณะนี้พบได้น้อย ไม่ใช่ในทุกเจเนอเรชั่น พบได้เท่าเทียมกันในทั้งเด็กชายและเด็กหญิง; 2) ลักษณะนี้สามารถปรากฏในเด็กได้แม้ว่าพ่อแม่จะไม่มีลักษณะนี้ก็ตาม 3) หากผู้ปกครองคนใดคนหนึ่งเป็นพาหะของลักษณะนี้ ก็จะไม่ปรากฏในเด็กหรือจะปรากฏในครึ่งหนึ่งของลูกหลาน
ประเภทของการสืบทอดลักษณะ 2. การถ่ายทอดทางพันธุกรรมแบบถอยอัตโนมัติ (Autosomal recessive inheritance) 1) ลักษณะนี้พบได้น้อย ไม่ใช่ในทุกเจเนอเรชั่น พบได้เท่าเทียมกันในทั้งเด็กชายและเด็กหญิง; 2) ลักษณะนี้สามารถปรากฏในเด็กได้แม้ว่าพ่อแม่จะไม่มีลักษณะนี้ก็ตาม 3) หากผู้ปกครองคนใดคนหนึ่งเป็นพาหะของลักษณะนี้ ก็จะไม่ปรากฏในเด็กหรือจะปรากฏในครึ่งหนึ่งของลูกหลาน
 ฟีนิลคีโตนูเรียคืออะไร? Phenylketonuria (PKU) เป็นโรคทางพันธุกรรมที่เพิ่มปริมาณกรดอะมิโนฟีนิลอะลานีนในเลือดให้อยู่ในระดับที่เป็นอันตราย (กรดอะมิโนเป็นหน่วยการสร้างของโปรตีน). หากไม่ได้รับการรักษา PKU ฟีนิลอะลานีนส่วนเกินอาจทำให้เกิดภาวะปัญญาอ่อนและปัญหาสุขภาพร้ายแรงอื่นๆ ได้ ผู้คนสืบทอด PKU ได้อย่างไร? PKU ได้รับการถ่ายทอดทางพันธุกรรมในลักษณะถอยอัตโนมัติ ซึ่งหมายความว่าจะต้องเปลี่ยนสำเนาของยีนสองชุดจึงจะได้รับผลกระทบจากโรคได้ บ่อยครั้งที่ผู้ปกครองของเด็กที่มีโรคถอยออโตโซมจะไม่ได้รับผลกระทบ แต่เป็นพาหะของยีนที่เปลี่ยนแปลงหนึ่งสำเนา
ฟีนิลคีโตนูเรียคืออะไร? Phenylketonuria (PKU) เป็นโรคทางพันธุกรรมที่เพิ่มปริมาณกรดอะมิโนฟีนิลอะลานีนในเลือดให้อยู่ในระดับที่เป็นอันตราย (กรดอะมิโนเป็นหน่วยการสร้างของโปรตีน). หากไม่ได้รับการรักษา PKU ฟีนิลอะลานีนส่วนเกินอาจทำให้เกิดภาวะปัญญาอ่อนและปัญหาสุขภาพร้ายแรงอื่นๆ ได้ ผู้คนสืบทอด PKU ได้อย่างไร? PKU ได้รับการถ่ายทอดทางพันธุกรรมในลักษณะถอยอัตโนมัติ ซึ่งหมายความว่าจะต้องเปลี่ยนสำเนาของยีนสองชุดจึงจะได้รับผลกระทบจากโรคได้ บ่อยครั้งที่ผู้ปกครองของเด็กที่มีโรคถอยออโตโซมจะไม่ได้รับผลกระทบ แต่เป็นพาหะของยีนที่เปลี่ยนแปลงหนึ่งสำเนา
 ประเภทของการสืบทอดลักษณะ 3. มรดกที่เชื่อมโยงกับเพศ: 1) X - มรดกที่โดดเด่น: ü ลักษณะนี้พบได้บ่อยในเพศหญิง; ü ถ้าแม่ป่วยและพ่อแข็งแรงดี ลักษณะนี้จะถ่ายทอดไปยังลูกหลานโดยไม่คำนึงถึงเพศ มันสามารถประจักษ์ได้ทั้งในเด็กหญิงและเด็กชาย ü ถ้าแม่แข็งแรงและพ่อป่วย ลูกสาวทุกคนก็จะแสดงอาการ แต่ลูกชายจะไม่แสดงอาการ
ประเภทของการสืบทอดลักษณะ 3. มรดกที่เชื่อมโยงกับเพศ: 1) X - มรดกที่โดดเด่น: ü ลักษณะนี้พบได้บ่อยในเพศหญิง; ü ถ้าแม่ป่วยและพ่อแข็งแรงดี ลักษณะนี้จะถ่ายทอดไปยังลูกหลานโดยไม่คำนึงถึงเพศ มันสามารถประจักษ์ได้ทั้งในเด็กหญิงและเด็กชาย ü ถ้าแม่แข็งแรงและพ่อป่วย ลูกสาวทุกคนก็จะแสดงอาการ แต่ลูกชายจะไม่แสดงอาการ
 3. มรดกทางเพศสัมพันธ์: 2) X - มรดกแบบด้อย: ลักษณะนี้มักพบในเพศชาย; บ่อยครั้งที่อาการนี้ปรากฏออกมาหลังจากผ่านไปหลายชั่วอายุคน หากทั้งพ่อและแม่มีสุขภาพดี แต่แม่เป็นเฮเทอโรไซกัส ลักษณะนี้มักจะปรากฏในลูกชาย 50% หากพ่อป่วยและแม่เป็นเฮเทอโรไซกัส ผู้หญิงก็สามารถมีลักษณะดังกล่าวได้เช่นกัน
3. มรดกทางเพศสัมพันธ์: 2) X - มรดกแบบด้อย: ลักษณะนี้มักพบในเพศชาย; บ่อยครั้งที่อาการนี้ปรากฏออกมาหลังจากผ่านไปหลายชั่วอายุคน หากทั้งพ่อและแม่มีสุขภาพดี แต่แม่เป็นเฮเทอโรไซกัส ลักษณะนี้มักจะปรากฏในลูกชาย 50% หากพ่อป่วยและแม่เป็นเฮเทอโรไซกัส ผู้หญิงก็สามารถมีลักษณะดังกล่าวได้เช่นกัน

 3. มรดกที่เชื่อมโยงกับเพศ: 3) มรดกที่เชื่อมโยงกับ Y: ลักษณะจะเกิดขึ้นเฉพาะในเพศชายเท่านั้น ถ้าพ่อมีคุณสมบัตินี้ ตามกฎแล้ว ลูกชายทุกคนก็มีคุณสมบัตินี้เช่นกัน
3. มรดกที่เชื่อมโยงกับเพศ: 3) มรดกที่เชื่อมโยงกับ Y: ลักษณะจะเกิดขึ้นเฉพาะในเพศชายเท่านั้น ถ้าพ่อมีคุณสมบัตินี้ ตามกฎแล้ว ลูกชายทุกคนก็มีคุณสมบัตินี้เช่นกัน
 ตัวอย่างการแก้ปัญหา โปรแบนด์เป็นผู้หญิงถนัดขวา พี่สาวสองคนของเธอถนัดขวา พี่ชายสองคนของเธอถนัดซ้าย แม่เป็นคนถนัดขวา เธอมีพี่ชายสองคนและน้องสาวหนึ่งคนถนัดขวาทั้งหมด ปู่และย่าเป็นคนถนัดขวา พ่อของโพรแบนด์ถนัดซ้าย พี่สาวและน้องชายของเขาถนัดซ้าย ส่วนพี่ชายและน้องสาวอีกสองคนถนัดขวา วิธีแก้ไข: 1. วาดสัญลักษณ์โปรแบนด์ เราแสดงการมีอยู่ของสัญญาณในโพรแบนด์
ตัวอย่างการแก้ปัญหา โปรแบนด์เป็นผู้หญิงถนัดขวา พี่สาวสองคนของเธอถนัดขวา พี่ชายสองคนของเธอถนัดซ้าย แม่เป็นคนถนัดขวา เธอมีพี่ชายสองคนและน้องสาวหนึ่งคนถนัดขวาทั้งหมด ปู่และย่าเป็นคนถนัดขวา พ่อของโพรแบนด์ถนัดซ้าย พี่สาวและน้องชายของเขาถนัดซ้าย ส่วนพี่ชายและน้องสาวอีกสองคนถนัดขวา วิธีแก้ไข: 1. วาดสัญลักษณ์โปรแบนด์ เราแสดงการมีอยู่ของสัญญาณในโพรแบนด์
 2. เราวางสัญลักษณ์พี่น้องของเธอไว้ข้างสัญลักษณ์โปรแบนด์ เราเชื่อมต่อพวกมันเข้ากับกราฟิคร็อคเกอร์
2. เราวางสัญลักษณ์พี่น้องของเธอไว้ข้างสัญลักษณ์โปรแบนด์ เราเชื่อมต่อพวกมันเข้ากับกราฟิคร็อคเกอร์




 7. กำหนดจีโนไทป์ของสมาชิกสายเลือด สัญลักษณ์แห่งการถนัดขวาปรากฏในทุกยุคทุกสมัยทั้งชายและหญิง สิ่งนี้บ่งชี้ถึงประเภทการสืบทอดลักษณะเด่นของออโตโซม ฉัน A- A- II A- A- A- Aa aa A- III aa Aa Aa A- aa
7. กำหนดจีโนไทป์ของสมาชิกสายเลือด สัญลักษณ์แห่งการถนัดขวาปรากฏในทุกยุคทุกสมัยทั้งชายและหญิง สิ่งนี้บ่งชี้ถึงประเภทการสืบทอดลักษณะเด่นของออโตโซม ฉัน A- A- II A- A- A- Aa aa A- III aa Aa Aa A- aa
 ภารกิจที่ 2 ตามสายเลือดที่แสดงในภาพ กำหนดลักษณะของการสำแดงลักษณะที่ระบุด้วยสีดำ (เด่น ถอย เชื่อมโยงกับเพศหรือไม่) กำหนดจีโนไทป์ของพ่อแม่และลูกในรุ่นแรก
ภารกิจที่ 2 ตามสายเลือดที่แสดงในภาพ กำหนดลักษณะของการสำแดงลักษณะที่ระบุด้วยสีดำ (เด่น ถอย เชื่อมโยงกับเพศหรือไม่) กำหนดจีโนไทป์ของพ่อแม่และลูกในรุ่นแรก
 แนวทางการแก้ปัญหา 1) ลักษณะด้อยไม่เกี่ยวเนื่องกับเพศ 2) จีโนไทป์ของพ่อแม่: แม่ - AA, พ่อ - AA หรือ Aa 3) จีโนไทป์ของลูก: ลูกชายและลูกสาวเฮเทอโรไซกัส - Aa
แนวทางการแก้ปัญหา 1) ลักษณะด้อยไม่เกี่ยวเนื่องกับเพศ 2) จีโนไทป์ของพ่อแม่: แม่ - AA, พ่อ - AA หรือ Aa 3) จีโนไทป์ของลูก: ลูกชายและลูกสาวเฮเทอโรไซกัส - Aa
 ภารกิจที่ 3 ใช้สายเลือดที่แสดงในแผนภาพ กำหนดประเภทและธรรมชาติของการสำแดงลักษณะที่เน้นด้วยสีดำ (เด่น ถอย เชื่อมโยงกับเพศหรือไม่ก็ได้) กำหนดจีโนไทป์ของเด็กในรุ่นแรก
ภารกิจที่ 3 ใช้สายเลือดที่แสดงในแผนภาพ กำหนดประเภทและธรรมชาติของการสำแดงลักษณะที่เน้นด้วยสีดำ (เด่น ถอย เชื่อมโยงกับเพศหรือไม่ก็ได้) กำหนดจีโนไทป์ของเด็กในรุ่นแรก
 แนวทางแก้ไขปัญหา 1) ลักษณะด้อย เชื่อมโยงกับโครโมโซม X 2) ลักษณะด้อย 2) จีโนไทป์ของพ่อแม่: แม่ – XHA, พ่อ – XAU; 3) จีโนไทป์ของเด็กใน F 1: ลูก - ฮา เอ่อ ลูกสาว - ฮ่าฮ่า ลูกสาว - ฮ่าฮ่า
แนวทางแก้ไขปัญหา 1) ลักษณะด้อย เชื่อมโยงกับโครโมโซม X 2) ลักษณะด้อย 2) จีโนไทป์ของพ่อแม่: แม่ – XHA, พ่อ – XAU; 3) จีโนไทป์ของเด็กใน F 1: ลูก - ฮา เอ่อ ลูกสาว - ฮ่าฮ่า ลูกสาว - ฮ่าฮ่า
 ภารกิจที่ 4 ใช้สายเลือดของบุคคลในภาพ กำหนดลักษณะการสืบทอดลักษณะ “ตาเล็ก” โดยเน้นด้วยสีดำ (เด่นหรือถอย เชื่อมโยงกับเพศหรือไม่ก็ได้) กำหนดจีโนไทป์ของพ่อแม่และลูกหลาน F 1 (1, 2, 3, 4, 5) 1 2 3 4 5
ภารกิจที่ 4 ใช้สายเลือดของบุคคลในภาพ กำหนดลักษณะการสืบทอดลักษณะ “ตาเล็ก” โดยเน้นด้วยสีดำ (เด่นหรือถอย เชื่อมโยงกับเพศหรือไม่ก็ได้) กำหนดจีโนไทป์ของพ่อแม่และลูกหลาน F 1 (1, 2, 3, 4, 5) 1 2 3 4 5
 แนวทางแก้ไขปัญหา 1) มีลักษณะด้อย ไม่เกี่ยวเนื่องกับเพศ 2) จีโนไทป์ของพ่อแม่: แม่ – Aa, พ่อ – Aa; 3) จีโนไทป์ของผู้สืบทอดใน F 1: 1, 2 – Aa, 3, 5 – AA หรือ Aa; 4 – อ๊า
แนวทางแก้ไขปัญหา 1) มีลักษณะด้อย ไม่เกี่ยวเนื่องกับเพศ 2) จีโนไทป์ของพ่อแม่: แม่ – Aa, พ่อ – Aa; 3) จีโนไทป์ของผู้สืบทอดใน F 1: 1, 2 – Aa, 3, 5 – AA หรือ Aa; 4 – อ๊า
 ตัวถอดรหัสองค์ประกอบเนื้อหาในชีววิทยา 3. 4 พันธุศาสตร์งานของมัน พันธุกรรมและความแปรปรวนเป็นคุณสมบัติของสิ่งมีชีวิต วิธีการทางพันธุศาสตร์ แนวคิดพื้นฐานทางพันธุกรรมและสัญลักษณ์ ทฤษฎีโครโมโซมของการถ่ายทอดทางพันธุกรรม แนวคิดสมัยใหม่เกี่ยวกับยีนและจีโนม 3. 5 รูปแบบของการถ่ายทอดทางพันธุกรรม, พื้นฐานทางเซลล์วิทยา รูปแบบของมรดกที่กำหนดโดย G. Mendel ซึ่งเป็นพื้นฐานทางเซลล์วิทยา (การผสมข้ามแบบโมโนและไดไฮบริด) กฎของมอร์แกน: การถ่ายทอดลักษณะที่เชื่อมโยงกัน การหยุดชะงักของการเชื่อมโยงของยีน พันธุศาสตร์ของเพศ การสืบทอดลักษณะที่เชื่อมโยงกับเพศ ปฏิสัมพันธ์ของยีน จีโนไทป์เป็นระบบบูรณาการ พันธุศาสตร์มนุษย์ วิธีการศึกษาพันธุศาสตร์มนุษย์ การแก้ปัญหาทางพันธุกรรม วาดโครงร่างการข้าม
ตัวถอดรหัสองค์ประกอบเนื้อหาในชีววิทยา 3. 4 พันธุศาสตร์งานของมัน พันธุกรรมและความแปรปรวนเป็นคุณสมบัติของสิ่งมีชีวิต วิธีการทางพันธุศาสตร์ แนวคิดพื้นฐานทางพันธุกรรมและสัญลักษณ์ ทฤษฎีโครโมโซมของการถ่ายทอดทางพันธุกรรม แนวคิดสมัยใหม่เกี่ยวกับยีนและจีโนม 3. 5 รูปแบบของการถ่ายทอดทางพันธุกรรม, พื้นฐานทางเซลล์วิทยา รูปแบบของมรดกที่กำหนดโดย G. Mendel ซึ่งเป็นพื้นฐานทางเซลล์วิทยา (การผสมข้ามแบบโมโนและไดไฮบริด) กฎของมอร์แกน: การถ่ายทอดลักษณะที่เชื่อมโยงกัน การหยุดชะงักของการเชื่อมโยงของยีน พันธุศาสตร์ของเพศ การสืบทอดลักษณะที่เชื่อมโยงกับเพศ ปฏิสัมพันธ์ของยีน จีโนไทป์เป็นระบบบูรณาการ พันธุศาสตร์มนุษย์ วิธีการศึกษาพันธุศาสตร์มนุษย์ การแก้ปัญหาทางพันธุกรรม วาดโครงร่างการข้าม
 ข้อมูลจำเพาะของข้อสอบทางชีววิทยา A 7. พันธุศาสตร์ หน้าที่ของมัน แนวคิดทางพันธุกรรมพื้นฐาน ก 8. รูปแบบการถ่ายทอดทางพันธุกรรม พันธุศาสตร์มนุษย์ ก 9. รูปแบบของความแปรปรวน ก 30. รูปแบบทางพันธุกรรม อิทธิพลของสารก่อกลายพันธุ์ต่ออุปกรณ์ทางพันธุกรรมของเซลล์และสิ่งมีชีวิต ค 6. การแก้ปัญหาทางพันธุศาสตร์เพื่อประยุกต์ความรู้ในสถานการณ์ใหม่
ข้อมูลจำเพาะของข้อสอบทางชีววิทยา A 7. พันธุศาสตร์ หน้าที่ของมัน แนวคิดทางพันธุกรรมพื้นฐาน ก 8. รูปแบบการถ่ายทอดทางพันธุกรรม พันธุศาสตร์มนุษย์ ก 9. รูปแบบของความแปรปรวน ก 30. รูปแบบทางพันธุกรรม อิทธิพลของสารก่อกลายพันธุ์ต่ออุปกรณ์ทางพันธุกรรมของเซลล์และสิ่งมีชีวิต ค 6. การแก้ปัญหาทางพันธุศาสตร์เพื่อประยุกต์ความรู้ในสถานการณ์ใหม่
 ส่วนที่ 1 พันธุศาสตร์มีความสำคัญอย่างยิ่งต่อการแพทย์ เนื่องจาก 1) ต่อสู้กับโรคระบาด 2) สร้างยาเพื่อรักษาผู้ป่วย 3) กำหนดสาเหตุของโรคทางพันธุกรรม 4) ปกป้องสิ่งแวดล้อมจากมลภาวะที่เกิดจากสารก่อกลายพันธุ์
ส่วนที่ 1 พันธุศาสตร์มีความสำคัญอย่างยิ่งต่อการแพทย์ เนื่องจาก 1) ต่อสู้กับโรคระบาด 2) สร้างยาเพื่อรักษาผู้ป่วย 3) กำหนดสาเหตุของโรคทางพันธุกรรม 4) ปกป้องสิ่งแวดล้อมจากมลภาวะที่เกิดจากสารก่อกลายพันธุ์
 2. วิธีที่ใช้ศึกษาลักษณะการแสดงลักษณะในพี่น้องที่พัฒนาจากไข่ที่ปฏิสนธิตัวเดียว เรียกว่า 1. 2. 3. 4. Hybridological Geneaological Cytogenetic Twin
2. วิธีที่ใช้ศึกษาลักษณะการแสดงลักษณะในพี่น้องที่พัฒนาจากไข่ที่ปฏิสนธิตัวเดียว เรียกว่า 1. 2. 3. 4. Hybridological Geneaological Cytogenetic Twin
 3. ใช้วิธีลำดับวงศ์ตระกูลเพื่อ 1) การได้รับยีนและการกลายพันธุ์ของจีโนม 2) การศึกษาอิทธิพลของการศึกษาต่อการสร้างเซลล์ของมนุษย์ 3) การวิจัยโรคทางพันธุกรรมของมนุษย์ 4) ศึกษาขั้นตอนวิวัฒนาการของโลกอินทรีย์
3. ใช้วิธีลำดับวงศ์ตระกูลเพื่อ 1) การได้รับยีนและการกลายพันธุ์ของจีโนม 2) การศึกษาอิทธิพลของการศึกษาต่อการสร้างเซลล์ของมนุษย์ 3) การวิจัยโรคทางพันธุกรรมของมนุษย์ 4) ศึกษาขั้นตอนวิวัฒนาการของโลกอินทรีย์
 4. การให้คำปรึกษาทางพันธุกรรมทางการแพทย์สำหรับคู่สมรสมีหน้าที่อะไร? 1. ระบุความโน้มเอียงของผู้ปกครองต่อโรคติดเชื้อ 2. กำหนดความเป็นไปได้ของการมีลูกแฝด 3. กำหนดโอกาสของโรคทางพันธุกรรมในเด็ก 4. ระบุความโน้มเอียงของผู้ปกครองต่อความผิดปกติของระบบเผาผลาญ
4. การให้คำปรึกษาทางพันธุกรรมทางการแพทย์สำหรับคู่สมรสมีหน้าที่อะไร? 1. ระบุความโน้มเอียงของผู้ปกครองต่อโรคติดเชื้อ 2. กำหนดความเป็นไปได้ของการมีลูกแฝด 3. กำหนดโอกาสของโรคทางพันธุกรรมในเด็ก 4. ระบุความโน้มเอียงของผู้ปกครองต่อความผิดปกติของระบบเผาผลาญ
 กำหนดจีโนไทป์ตามฟีโนไทป์ สีตาในบุคคลถูกกำหนดโดยยีนออโตโซม ตาบอดสีเป็นยีนด้อยที่เชื่อมโยงกับเพศ ตรวจจีโนไทป์ของผู้หญิงตาสีน้ำตาลที่มีการมองเห็นสีปกติ ซึ่งพ่อตาบอดสี (ตาสีน้ำตาลเด่นกว่าตาสีฟ้า) 1) AAXDXD 3) Aa. Xd 2) อ๋อ XDXd 4) อ๋อ XDXd
กำหนดจีโนไทป์ตามฟีโนไทป์ สีตาในบุคคลถูกกำหนดโดยยีนออโตโซม ตาบอดสีเป็นยีนด้อยที่เชื่อมโยงกับเพศ ตรวจจีโนไทป์ของผู้หญิงตาสีน้ำตาลที่มีการมองเห็นสีปกติ ซึ่งพ่อตาบอดสี (ตาสีน้ำตาลเด่นกว่าตาสีฟ้า) 1) AAXDXD 3) Aa. Xd 2) อ๋อ XDXd 4) อ๋อ XDXd
 ภาค ค การแก้ปัญหาทางพันธุกรรมเรื่องการประยุกต์ใช้ความรู้ในสถานการณ์ใหม่ การผสมข้ามพันธุ์แบบไดไฮบริด การสืบทอดลักษณะที่เชื่อมโยงทางเพศ การถ่ายทอดลักษณะที่เชื่อมโยงกัน (แบบมีข้าม และไม่ข้าม) การกำหนดกลุ่มเลือด การวิเคราะห์สายเลือด
ภาค ค การแก้ปัญหาทางพันธุกรรมเรื่องการประยุกต์ใช้ความรู้ในสถานการณ์ใหม่ การผสมข้ามพันธุ์แบบไดไฮบริด การสืบทอดลักษณะที่เชื่อมโยงทางเพศ การถ่ายทอดลักษณะที่เชื่อมโยงกัน (แบบมีข้าม และไม่ข้าม) การกำหนดกลุ่มเลือด การวิเคราะห์สายเลือด
 ส่วน C ในมนุษย์ การถ่ายทอดทางพันธุกรรมของโรคเผือกไม่เกี่ยวข้องกับเพศ (A - การมีอยู่ของเมลานินในเซลล์ผิวหนัง และ - การไม่มีเมลานินในเซลล์ผิวหนัง - โรคเผือก) และฮีโมฟีเลียมีความเชื่อมโยงทางเพศ (XH - การแข็งตัวของเลือดปกติ , Xh - ฮีโมฟีเลีย) พิจารณาจีโนไทป์ของพ่อแม่ รวมถึงจีโนไทป์ที่เป็นไปได้ เพศ และฟีโนไทป์ของเด็กจากการแต่งงานของหญิงประเภทไดโฮโมไซกัส ซึ่งเป็นเรื่องปกติสำหรับอัลลีลทั้งสอง และชายเผือกที่เป็นโรคฮีโมฟีเลีย สร้างไดอะแกรมสำหรับการแก้ปัญหา
ส่วน C ในมนุษย์ การถ่ายทอดทางพันธุกรรมของโรคเผือกไม่เกี่ยวข้องกับเพศ (A - การมีอยู่ของเมลานินในเซลล์ผิวหนัง และ - การไม่มีเมลานินในเซลล์ผิวหนัง - โรคเผือก) และฮีโมฟีเลียมีความเชื่อมโยงทางเพศ (XH - การแข็งตัวของเลือดปกติ , Xh - ฮีโมฟีเลีย) พิจารณาจีโนไทป์ของพ่อแม่ รวมถึงจีโนไทป์ที่เป็นไปได้ เพศ และฟีโนไทป์ของเด็กจากการแต่งงานของหญิงประเภทไดโฮโมไซกัส ซึ่งเป็นเรื่องปกติสำหรับอัลลีลทั้งสอง และชายเผือกที่เป็นโรคฮีโมฟีเลีย สร้างไดอะแกรมสำหรับการแก้ปัญหา
 โครงการแก้ไขปัญหาประกอบด้วย: 1) จีโนไทป์ของผู้ปกครอง: aniAAXHXH (AXH gametes); ♂อ่า. Xh. Y (เซลล์สืบพันธุ์ a. Xh, a. Y); 2) จีโนไทป์และเพศของเด็ก: 🙋Aa. XHXH; ♂อ๋า. เอ็กซ์ไฮ; 3) ฟีโนไทป์ของเด็ก: เด็กผู้หญิงที่ภายนอกปกติสำหรับอัลลีลทั้งสอง แต่เป็นพาหะของยีนสำหรับโรคเผือกและโรคฮีโมฟีเลีย เด็กผู้ชายที่ภายนอกดูปกติสำหรับอัลลีลทั้งสอง แต่เป็นพาหะของยีนเผือก
โครงการแก้ไขปัญหาประกอบด้วย: 1) จีโนไทป์ของผู้ปกครอง: aniAAXHXH (AXH gametes); ♂อ่า. Xh. Y (เซลล์สืบพันธุ์ a. Xh, a. Y); 2) จีโนไทป์และเพศของเด็ก: 🙋Aa. XHXH; ♂อ๋า. เอ็กซ์ไฮ; 3) ฟีโนไทป์ของเด็ก: เด็กผู้หญิงที่ภายนอกปกติสำหรับอัลลีลทั้งสอง แต่เป็นพาหะของยีนสำหรับโรคเผือกและโรคฮีโมฟีเลีย เด็กผู้ชายที่ภายนอกดูปกติสำหรับอัลลีลทั้งสอง แต่เป็นพาหะของยีนเผือก
ความผิดปกติที่นำไปสู่ระดับที่เพิ่มขึ้น ฟีนิลอะลานีนเลือด ซึ่งส่วนใหญ่มักขาดฟีนิลอะลานีนไฮดรอกซีเลส (PAH) หรือฟีนิลคีโตนูเรีย (PKU) แสดงให้เห็นหลักการเกือบทั้งหมดของพันธุศาสตร์ทางชีวเคมีที่เกี่ยวข้องกับข้อบกพร่องของเอนไซม์ ความผิดปกติทางพันธุกรรมทั้งหมดของการเผาผลาญฟีนิลอะลานีนเป็นผลมาจากการกลายพันธุ์ที่สูญเสียการทำงานในยีนที่เข้ารหัส PAH หรือในยีนที่จำเป็นสำหรับการสังเคราะห์หรือการฟื้นฟูโคแฟกเตอร์ BH4
ฟีนิลคีโตนูเรียแบบคลาสสิก(PKU) ได้รับการพิจารณาอย่างถูกต้องว่าเป็นตัวแทนที่เป็นแบบอย่างของข้อผิดพลาดโดยกำเนิดของการเผาผลาญ เป็นโรคที่เกิดจากการสลายตัวของฟีนิลอะลานีนแบบถอยอัตโนมัติที่เกิดจากการกลายพันธุ์ของยีนที่เข้ารหัส PAH ซึ่งเป็นเอนไซม์ที่เปลี่ยนฟีนิลอะลานีนเป็นไทโรซีน การค้นพบฟีนิลคีโตนูเรีย (PKU) ของ Fehling ในปี พ.ศ. 2477 ถือเป็นการค้นพบครั้งแรกที่แสดงให้เห็นถึงความบกพร่องทางพันธุกรรมอันเป็นสาเหตุของภาวะปัญญาอ่อน
เนื่องจากไม่สามารถรีไซเคิลได้ ฟีนิลอะลานีนผู้ป่วยที่เป็นโรคฟีนิลคีโตนูเรีย (PKU) จะสะสมกรดอะมิโนนี้ในของเหลวในร่างกาย ภาวะไฮเปอร์ฟีนิลอะลานินีเมียทำลายระบบประสาทส่วนกลางที่กำลังพัฒนาในวัยเด็ก และรบกวนการทำงานของสมองผู้ใหญ่ ฟีนิลอะลานีนส่วนเล็กๆ จะถูกเผาผลาญโดยวิถีทางอื่น ทำให้เกิดกรดฟีนิลไพรูวิก (กรดคีโตที่เป็นชื่อของโรค) ในปริมาณเพิ่มขึ้น และสารอื่นๆ จะถูกขับออกทางปัสสาวะ
แม้ว่าจะเป็นที่น่าสนใจก็ตาม ข้อบกพร่องของเอนไซม์เป็นที่รู้กันมานานหลายทศวรรษแล้ว แต่ยังไม่ทราบกลไกการเกิดโรคที่แน่นอนของการเพิ่มขึ้นของฟีนิลอะลานีนที่ทำลายสมอง ที่สำคัญ การพัฒนาความเสียหายทางระบบประสาทที่เกิดจากบล็อกการเผาผลาญใน PKU แบบคลาสสิกส่วนใหญ่สามารถป้องกันได้โดยการเปลี่ยนแปลงอาหารที่ป้องกันการสะสมฟีนิลอะลานีน การรักษาภาวะฟีนิลคีโตนูเรีย (PKU) ได้กลายเป็นต้นแบบในการรักษาโรคทางเมตาบอลิซึมหลายชนิด ผลลัพธ์ที่อาจได้รับการปรับปรุงให้ดีขึ้นโดยการป้องกันการสะสมของสารตั้งต้นของเอนไซม์และอนุพันธ์ของเอนไซม์
การตรวจคัดกรองภาวะฟีนิลคีโตนูเรีย (PKU) ทารกแรกเกิด
ประชากรมีการใช้กันอย่างแพร่หลาย การคัดกรองทารกแรกเกิดสำหรับภาวะฟีนิลคีโตนูเรีย (PKU) Phenylketonuria (PKU) เป็นตัวอย่างของโรคทางพันธุกรรมที่รับประกันการตรวจคัดกรองทารกแรกเกิดจำนวนมาก โรคนี้พบได้บ่อยในหลายประชากร (มากถึง 1 ใน 2,900 ทารกแรกเกิดที่ยังมีชีวิตอยู่) การรักษาที่เริ่มต้นตั้งแต่อายุยังน้อยมีประสิทธิผลมาก หากไม่ได้รับการรักษา ภาวะปัญญาอ่อนขั้นรุนแรงจะเกิดขึ้นอย่างหลีกเลี่ยงไม่ได้ การตรวจคัดกรองจะดำเนินการไม่กี่วันหลังคลอด
เลือดหยดหนึ่งที่ได้จากการเจาะ ส้นเท้านำไปใช้กับกระดาษกรอง ตากแห้ง และส่งไปยังห้องปฏิบัติการส่วนกลางเพื่อประเมินระดับฟีนิลอะลานีนในเลือดและอัตราส่วนฟีนิลอะลานีน/ไทโรซีน ในอดีตจะมีการเก็บตัวอย่างก่อนที่ทารกจะออกจากโรงพยาบาล แนวโน้มการจำหน่ายแม่และทารกแรกเกิดหลังคลอดก่อนกำหนดได้เปลี่ยนแปลงแนวทางปฏิบัตินี้ ไม่ควรทำการทดสอบก่อนอายุ 24 ชั่วโมง เนื่องจากระดับฟีนิลอะลานีนในฟีนิลคีโตนูเรีย (PKU) จะไม่เพิ่มขึ้นจนกระทั่งหลังคลอด ผลการทดสอบที่เป็นบวกควรได้รับการยืนยันอย่างรวดเร็ว เนื่องจากการชะลอการเริ่มต้นการรักษานานกว่า 4 สัปดาห์หลังคลอดไม่ได้หลีกเลี่ยงผลกระทบต่อสถานะทางปัญญาของผู้ป่วยโรคฟีนิลคีโตนูเรีย (PKU)
รูปแบบต่างๆ ของฟีนิลคีโตนูเรียและภาวะไฮเปอร์ฟีนิลอะลานิเมีย
เนื่องจาก (PKU) เกี่ยวข้องกับการขาดอย่างรุนแรงของการทำงานของฟีนิลอะลานีนไฮดรอกซีเลส (PAH) (น้อยกว่า 1% เมื่อเทียบกับกลุ่มควบคุม) PAH กลายพันธุ์ที่มีฤทธิ์ตกค้างจะทำให้เกิดอาการทางฟีโนไทป์ที่รุนแรงน้อยกว่า ที่เรียกว่าภาวะฟีนิลอะลานินในเลือดสูง และฟีนิลคีโตนูเรีย (PKU) ผิดปรกติ
Hyperphenylalaninemia phenylketonuria (PKU) นอกเหนือจาก phenylketonuria (PKU) ได้รับการวินิจฉัยว่าความเข้มข้นของฟีนิลอะลานีนในพลาสมาต่ำกว่า 1 มิลลิโมลต่อลิตรในการรับประทานอาหารตามปกติ ระดับของภาวะฟีนิลอะลานินในเลือดสูงระดับนี้สูงกว่าปกติเพียง 10 เท่า และต่ำกว่าความเข้มข้นที่พบในโรคฟีนิลคีโตนูเรียแบบดั้งเดิม (PKU) (>1 มิลลิโมล/ลิตร) อย่างมีนัยสำคัญ การเพิ่มขึ้นปานกลางของฟีนิลอะลานีนในภาวะฟีนิลอะลานินในเลือดสูงไม่น่าจะเป็นอันตรายต่อการทำงานของสมอง และอาจมีประโยชน์ด้วยซ้ำหากการเพิ่มขึ้นเล็กน้อย (<0,4 ммоль), такие дети обращают на себя внимание врачей только благодаря скринингу. Их нормальный фенотип оказался наилучшим показателем безопасного уровня фенилаланина плазмы, который не следует превышать при лечении пациентов с классической фенилкетонурии (ФКУ).
ผิดปกติ(PKU) - หมวดหมู่ที่รวมถึงผู้ป่วยที่มีระดับฟีนิลอะลานีนที่อยู่ตรงกลางระหว่าง PKU แบบคลาสสิกและภาวะฟีนิลอะลานีนในเลือดสูง ผู้ป่วยดังกล่าวจำเป็นต้องมีการจำกัดฟีนิลอะลานีนในอาหาร แต่น้อยกว่าผู้ป่วยที่มีภาวะฟีนิลคีโตนูเรียแบบดั้งเดิม (PKU) ความซับซ้อนของฟีโนไทป์ทางคลินิกทั้งสามชนิดนี้ที่มีการกลายพันธุ์ในยีน PAH เป็นตัวอย่างของความแตกต่างทางคลินิก
Hyperphenylalaninemia: ความหลากหลายของอัลลีลิกและโลคัสในฟีนิลคีโตนูเรีย (PKU)
โมเลกุล ข้อบกพร่องในยีนฟีนิลอะลานีน ไฮดรอกซีเลส ผู้ป่วยที่มีภาวะไฮเปอร์ฟีนิลอะลานินในเลือดสูง รวมถึงคลาสสิกฟีนิลคีโตนูเรีย (PKU), ฟีนิลคีโตนูเรียผิดปกติ (PKU) และภาวะฟีนิลอะลานินในเลือดสูงที่ไม่เป็นอันตราย แสดงระดับความแตกต่างของอัลลีลิกที่โดดเด่นที่ตำแหน่งฟีนิลอะลานีนไฮดรอกซีเลส (PAH) (การกลายพันธุ์ที่แตกต่างกันมากกว่า 400 รายการทั่วโลก)
อัลลีลส่วนใหญ่ ฟีนิลอะลานีน ไฮดรอกซีเลส(PAH) เป็นการกลายพันธุ์ที่ค่อนข้างหายากซึ่งรบกวนคุณสมบัติของเอนไซม์ของฟีนิลอะลานีนไฮดรอกซีเลส (PAH) และนำไปสู่ภาวะไฮเปอร์ฟีนิลอะลานิเมีย แม้ว่าจะพบความหลากหลายที่เป็นพิษเป็นภัยหรือตัวแปรที่ไม่เป็นพิษเป็นภัยที่พบได้น้อยกว่าก็ตาม
ในประชากร เชื้อสายยุโรปประมาณสองในสามของโครโมโซมกลายพันธุ์ที่รู้จักจะแสดงด้วยการกลายพันธุ์หกครั้ง การกลายพันธุ์อีก 6 แบบมีส่วนทำให้เกิดการกลายพันธุ์ของฟีนิลอะลานีนไฮดรอกซีเลส (PAH) มากกว่า 80% ในประชากรเอเชีย การกลายพันธุ์ที่ทำให้เกิดโรคอื่น ๆ นั้นพบได้น้อย เพื่อให้ข้อมูลนี้เข้าถึงได้อย่างกว้างขวาง สมาคมระหว่างประเทศได้พัฒนาฐานข้อมูลการกลายพันธุ์ในยีนฟีนิลอะลานีนไฮดรอกซีเลส (PAH)
ทั้งหมด ประชากรมีความแตกต่างทางพันธุกรรมที่ชัดเจนของฟีนิลอะลานีนไฮดรอกซีเลส (PAH) เนื่องจากความหลากหลายของอัลลีลในระดับที่สูง ผู้ป่วยส่วนใหญ่ที่มีฟีนิลคีโตนูเรีย (PKU) ในประชากรจำนวนมากจึงเป็นเฮเทอโรไซโกตแบบผสม (นั่นคือ พวกมันมีอัลลีลที่ทำให้เกิดโรคที่แตกต่างกันสองชนิด) ซึ่งสอดคล้องอย่างสมบูรณ์กับความหลากหลายของเอนไซม์และฟีโนไทป์ที่สังเกตได้ใน ความผิดปกติของฟีนิลอะลานีนไฮดรอกซีเลส (PAH)

ในตอนแรกดูเหมือนว่าความรู้เกี่ยวกับจีโนไทป์นั้น ฟีนิลอะลานีน ไฮดรอกซีเลส(FA) ทำนายรายละเอียดฟีโนไทป์ได้อย่างน่าเชื่อถือ ความคาดหวังนี้ไม่ได้รับการพิสูจน์อย่างสมบูรณ์ แม้ว่าจะพบความสัมพันธ์บางอย่างระหว่างจีโนไทป์ PAH และฟีโนไทป์ทางชีวเคมีก็ตาม
โดยทั่วไป การกลายพันธุ์ที่ระงับหรือลดกิจกรรมลงอย่างมาก ฟีนิลอะลานีน ไฮดรอกซีเลส(PAH) ทำให้เกิดฟีนิลคีโตนูเรียแบบดั้งเดิม (PKU) ในขณะที่การกลายพันธุ์ที่ทำให้เกิดการทำงานของเอนไซม์ที่ตกค้างในปริมาณมากเพียงพอนั้นสัมพันธ์กับฟีโนไทป์ที่ไม่รุนแรง
อย่างไรก็ตามมีการกลายพันธุ์บ้าง ฟีนิลอะลานีน ไฮดรอกซีเลส(FA) ในผู้ป่วยโฮโมไซกัสจะกำหนดสเปกตรัมของฟีโนไทป์ทั้งหมด ตั้งแต่ฟีนิลคีโตนูเรียแบบคลาสสิก (PKU) ไปจนถึงภาวะฟีนิลอะลานินในเลือดสูงที่ไม่เป็นอันตราย
ดังนั้นจึงเห็นได้ชัดเจนว่าในรูปแบบนั้น ฟีโนไทป์จากการสังเกตพบในจีโนไทป์ที่เฉพาะเจาะจง ปัจจัยทางชีววิทยาอื่นๆ ที่ไม่สามารถระบุได้ก็มีส่วนเกี่ยวข้อง รวมถึงยีนตัวดัดแปลงอย่างไม่ต้องสงสัย การสังเกตนี้ ซึ่งขณะนี้ได้รับการยอมรับว่าเป็นลักษณะทั่วไปของโรคที่ทำให้เกิดโมโนเจนิกหลายชนิด บ่งชี้ว่าแม้แต่โรคที่ทำให้เกิดโมโนเจนิก เช่น ฟีนิลคีโตนูเรีย (PKU) ก็ไม่ใช่โรคที่เกิดจากพันธุกรรมอย่างง่าย
ข้อบกพร่องในการเผาผลาญ tetrahydrobiopterin ใน phenylketonuria (PKU)
ในตอนแรกเชื่อกันว่าเด็กทุกคนที่มีกรรมพันธุ์ ภาวะไฮเปอร์ฟีนิลอะลานีเมียมีภาวะขาดฟีนิลอะลานีนไฮดรอกซีเลส (PAH) หลัก ขณะนี้เป็นที่ชัดเจนว่าผู้ป่วยประมาณ 1-3% มียีน PAH ปกติ และภาวะฟีนิลอะลานินในเลือดสูงเป็นผลมาจากความบกพร่องทางพันธุกรรมในยีนตัวใดตัวหนึ่งจากยีนอื่นๆ ที่เกี่ยวข้องกับการสังเคราะห์หรือการงอกใหม่ของโคแฟกเตอร์ PAH หรือ BH4 การเชื่อมโยงกันของฟีโนไทป์ประเภทหนึ่ง เช่น ภาวะไฮเปอร์ฟีนิลอะลานินีเมีย กับการกลายพันธุ์ในยีนที่ต่างกัน เป็นตัวอย่างหนึ่งของความแตกต่างของโลคัส
ดังที่แสดงโดยการกลายพันธุ์ของยีนเข้ารหัสโปรตีน ฟีนิลอะลานีน ไฮดรอกซีเลส(PAH) และเมแทบอลิซึมของโคแฟกเตอร์ไบโอพเทอริน ซึ่งเป็นโปรตีนที่ถูกเข้ารหัสโดยยีนที่แสดงความแตกต่างของโลคัส มักจะมีส่วนร่วมในปฏิกิริยาทางชีวเคมีสายโซ่เดียวกัน ผู้ป่วยที่มีภาวะขาด BH4 ได้รับการระบุเป็นครั้งแรกเนื่องจากแม้จะรักษาความเข้มข้นของฟีนิลอะลานีนในอาหารให้ต่ำได้สำเร็จ แต่พวกเขาก็เริ่มมีปัญหาทางระบบประสาทอย่างลึกซึ้งตั้งแต่เริ่มแรก
มีการอธิบายผลลัพธ์ที่ไม่ดีบางส่วน ความต้องการโคแฟคเตอร์ BH4สำหรับการทำงานของเอนไซม์อีก 2 ชนิด ได้แก่ ไทโรซีน ไฮดรอกซีเลส และทริปโตเฟน ไฮดรอกซีเลส ไฮดรอกซีเลสทั้งสองนี้มีความสำคัญอย่างยิ่งต่อการสังเคราะห์สารสื่อประสาทโมโนเอมีน เช่น ดีไฮดรอกซีฟีนิลอะลานีน, นอร์เอพิเนฟริน, อะดรีนาลีน และเซโรโทนิน ผู้ป่วยที่มีภาวะพร่อง BH4 มีความบกพร่องในการสังเคราะห์ทางชีวภาพจาก GTP หรือการสร้าง BH4 ใหม่ เช่นเดียวกับโรคฟีนิลคีโตนูเรีย (PKU) แบบคลาสสิก ความผิดปกตินี้ถ่ายทอดทางพันธุกรรมในลักษณะถอยแบบออโตโซม
สิ่งสำคัญคือต้องแยกแยะผู้ป่วยที่มีความบกพร่องในการเผาผลาญ BH4 จากผู้ป่วยที่มีการกลายพันธุ์มา ฟีนิลอะลานีน ไฮดรอกซีเลส(FA) เนื่องจากการรักษาแตกต่างกันอย่างเห็นได้ชัด ประการแรก เนื่องจากโครงสร้างโปรตีนของฟีนิลอะลานีนไฮดรอกซีเลส (PAH) เป็นเรื่องปกติในผู้ป่วยที่มีความผิดปกติของ BH4 กิจกรรมของมันอาจกลับคืนมาได้หากผู้ป่วยเหล่านี้ได้รับ BH4 ในปริมาณมาก ซึ่งทำให้ระดับฟีนิลอะลานีนในพลาสมาลดลง ดังนั้น ระดับของการจำกัดฟีนิลอะลานีนในอาหารของผู้ป่วยที่มีความบกพร่องในการเผาผลาญ BH4 จึงสามารถลดลงได้อย่างมีนัยสำคัญ และผู้ป่วยบางรายสามารถเปลี่ยนมารับประทานอาหารตามปกติได้ (กล่าวคือ โดยไม่มีข้อจำกัดฟีนิลอะลานีน)
ประการที่สองคุณต้องลองด้วย ทำให้เป็นปกติระดับสารสื่อประสาทในสมองของผู้ป่วยเหล่านี้โดยการบริหารผลิตภัณฑ์ tyrosine hydroxylase และ tryptophan hydroxylase: L-dopa และ 5-hydroxytryptophan ตามลำดับ ด้วยเหตุผลเหล่านี้ ทารกแรกเกิดทุกคนที่มีภาวะฟีนิลอะลานินในเลือดสูงควรได้รับการประเมินความผิดปกติในการเผาผลาญ BH4
ปฏิกิริยาต่อ tetrahydrobiopterin ที่มีการกลายพันธุ์ในยีน PAH ใน phenylketonuria (PKU)
ในผู้ป่วยส่วนใหญ่จะมีการกลายพันธุ์ของยีน ฟีนิลอะลานีน ไฮดรอกซีเลส(PAH) และไม่ได้อยู่ในการเผาผลาญของ BH4 ระดับฟีนิลอะลานีนในเลือดลดลงอย่างเห็นได้ชัดในระหว่างการรับประทานโคแฟคเตอร์ฟีนิลอะลานีนไฮดรอกซีเลส (PAH) BH4 ในปริมาณมากทางปาก ผู้ป่วยที่มีฤทธิ์ฟีนิลอะลานีนไฮดรอกซีเลส (PAH) ตกค้างอย่างมีนัยสำคัญ (เช่น ผู้ป่วยที่มีภาวะฟีนิลคีโตนูเรีย (PKU) ผิดปรกติและภาวะฟีนิลอะลานินในเลือดสูง) ตอบสนองต่อการรักษาดังกล่าวได้ดีที่สุด แต่มีผู้ป่วยจำนวนเล็กน้อยถึงแม้จะเป็นโรคฟีนิลคีโตนูเรียแบบคลาสสิก (PKU) ก็ตอบสนองต่อการรักษานี้เช่นกัน ในเวลาเดียวกัน การมีอยู่ของกิจกรรม PAH ที่ตกค้างไม่ได้รับประกันผลต่อระดับฟีนิลอะลานีนในพลาสมาเมื่อมีการกำหนด BH4
เป็นไปได้มากว่าระดับของการตอบสนอง ปฏิกิริยาบน BH4 ขึ้นอยู่กับคุณสมบัติเฉพาะของโปรตีนกลายพันธุ์ฟีนิลอะลานีนไฮดรอกซีเลส (PAH) แต่ละตัว ซึ่งสะท้อนถึงความหลากหลายของอัลลีลที่เป็นพื้นฐานของการกลายพันธุ์ของ PAH แสดงให้เห็นว่าการนำ BH4 เข้าสู่อาหารมีผลการรักษาผ่านกลไกหลายประการที่เกิดจากการเพิ่มปริมาณของโคแฟกเตอร์ปกติที่สัมผัสกับการกลายพันธุ์
กลไกเหล่านี้รวมถึงการรักษาเสถียรภาพของสายพันธุ์กลาย เอนไซม์การป้องกันเอนไซม์จากการเสื่อมสลายของเซลล์ การเพิ่มโคแฟกเตอร์ให้กับเอนไซม์ที่มีความสัมพันธ์ต่ำกับ BH4 และผลประโยชน์อื่นๆ ในคุณสมบัติทางจลน์และการเร่งปฏิกิริยาของเอนไซม์ การให้โคแฟกเตอร์ในปริมาณที่เพิ่มขึ้นเป็นกลยุทธ์ทั่วไปที่ใช้ในการรักษาข้อผิดพลาดแต่กำเนิดของการเผาผลาญ
การกลายพันธุ์ของยีนจำนวนหนึ่ง ซึ่งมีการเปลี่ยนแปลงโครงสร้างของยีนเพียงยีนเดียว ทำให้เกิดภาวะปัญญาอ่อน ตามการประมาณการใน 7-10% ของผู้ป่วยที่มี oligophrenia มีสาเหตุมาจากการกลายพันธุ์ประเภทนี้
ชุดของปฏิกิริยาทางชีวเคมีที่เกิดขึ้นในร่างกายเรียกว่าเมแทบอลิซึม ยีนจำนวนมากเข้ารหัสโปรตีนที่มีส่วนร่วมเป็นเอนไซม์ในปฏิกิริยาเมแทบอลิซึมบางอย่าง การกลายพันธุ์ในยีนดังกล่าวอาจทำให้ร่างกายผลิตเอนไซม์ที่ทำงานน้อยลงหรือไม่ทำงานโดยสิ้นเชิง และบางครั้งอาจนำไปสู่การหยุดการสังเคราะห์เอนไซม์โดยสมบูรณ์ ในกรณีนี้ปฏิกิริยาที่ปกติดำเนินการโดยเอนไซม์นี้อาจช้าลงหรือไม่เกิดขึ้นเลยซึ่งทำให้เกิดความผิดปกติทางพันธุกรรมที่เกี่ยวข้อง - หนึ่งในสิ่งที่เรียกว่าข้อผิดพลาดโดยกำเนิดของการเผาผลาญ โรคทางพันธุกรรมทางพันธุกรรมที่พบบ่อยที่สุด ได้แก่ ฟีนิลคีโตนูเรีย โรคโลหิตจางชนิดเคียว โรคเทย์-แซคส์ โรคฮีโมฟีเลีย และเบาหวาน ขอบเขตที่พวกมันมีอิทธิพลต่อฟีโนไทป์นั้นขึ้นอยู่กับความสำคัญของเอนไซม์ที่ได้รับผลกระทบต่อสิ่งมีชีวิต เราเห็นข้างต้นว่าโรค Tay-Sachs และโรคซิสติกไฟโบรซิสทำให้เสียชีวิตได้ ความผิดปกติทางพันธุกรรมอื่นๆ ทำให้เกิดปัญหาร้ายแรงต่างๆ ในร่างกาย แต่ก็ไม่ร้ายแรงถึงชีวิต
Phenylketonuria และ albinism ส่งผลต่อวิถีเมแทบอลิซึมเดียวกัน
Phenylketonuria เป็นโรคที่โครงสร้างของเอนไซม์ที่เกี่ยวข้องกับการเผาผลาญของกรดอะมิโนฟีนิลอะลานีน (ฟีนิลอะลานีนไฮดรอกซีเลส) ถูกทำลายซึ่งเป็นผลมาจากการกลายพันธุ์ เอนไซม์นี้จำเป็นสำหรับการเปลี่ยนฟีนิลอะลานีนเป็นไทโรซีน โรคประเภทนี้เรียกว่าเอนไซม์พาธีย์เช่น เกิดจากความบกพร่องของเอนไซม์ ด้วยโรคนี้ฟีนิลอะลานีนและผลิตภัณฑ์จากการเผาผลาญที่ไม่เหมาะสม (กรดฟีนิลอะซิติก) จะสะสมในเลือดซึ่งนำไปสู่ความเสียหายต่อระบบประสาทที่กำลังพัฒนา นี่คือการทำลายไมอีลินและความเสื่อมของระบบประสาทสปองจิฟอร์มเป็นหลัก ภาวะปัญญาอ่อน ภาวะศีรษะเล็ก โรคจิต อาการสั่น อาการชัก และอาการเกร็งเกิดขึ้น
โรคฟีนิลคีโตนูเรียส่งผลกระทบต่อบุคคลที่เป็นโฮโมไซกัสสำหรับยีนด้อยซึ่งทำให้ไม่สามารถสังเคราะห์เอนไซม์ตัวใดตัวหนึ่งที่จำเป็นในการเปลี่ยนกรดอะมิโนฟีนิลอะลานีนให้เป็นกรดอะมิโนตัวอื่นซึ่งก็คือไทโรซีน แทนที่จะเปลี่ยนเป็นไทโรซีน ฟีนิลอะลานีนจะถูกเปลี่ยนเป็นกรดฟีนิลไพรูวิก ซึ่งสะสมในปริมาณที่เป็นพิษในเลือด ส่งผลต่อสมอง และ (หากไม่ได้รับการรักษาอย่างทันท่วงที) ทำให้เกิดภาวะปัญญาอ่อน ปัสสาวะของผู้ป่วยยังมีกรดฟีนิลไพรูวิกซึ่งทำให้มีกลิ่นเฉพาะตัว ปัจจุบัน ฟีนิลคีโตนูเรียได้รับการรักษาด้วยการรับประทานอาหารพิเศษ ในการทำเช่นนี้ในปีแรกของชีวิตของเด็ก ฟีนิลอะลานีนจะถูกแยกออกจากอาหารของเขาเกือบทั้งหมด เมื่อการพัฒนาสมองเสร็จสมบูรณ์ ผู้ป่วยที่มีภาวะฟีนิลคีโตนูเรียจะต้องรับประทานอาหารตามปกติ แต่ผู้หญิงที่มีความผิดปกติทางพันธุกรรมนี้ควรรับประทานอาหารที่มีฟีนิลอะลานีนต่ำในระหว่างตั้งครรภ์ เพื่อป้องกันการพัฒนาที่ผิดปกติของสมองของทารกในครรภ์ ในสหรัฐอเมริกา ในหลายรัฐ ทารกแรกเกิดทุกคนจะต้องผ่านการทดสอบพิเศษสำหรับ PKU และข้อผิดพลาดทางเมตาบอลิซึมแต่กำเนิดอื่นๆ
บุคคลที่มีลักษณะโฮโมไซกัสสำหรับยีนเผือกขาดเอนไซม์ที่ปกติจะกระตุ้นการเปลี่ยนไทโรซีนไปเป็นเมลานิน กล่าวคือ เม็ดสีที่กำหนดสีน้ำตาลหรือสีดำของดวงตา ผม และผิวหนัง อัลบีโนสมีผมสีขาว ผิวและดวงตาที่สว่างมาก โดยธรรมชาติแล้ว คำถามอาจเกิดขึ้นได้ว่าผู้ป่วยที่เป็นโรคฟีนิลคีโตนูเรียเป็นโรคเผือกหรือไม่ เนื่องจากร่างกายของพวกเขาไม่ได้ผลิตไทโรซีน ซึ่งในที่สุดจะผลิตเมลานินขึ้นมา อย่างไรก็ตามผู้ป่วยดังกล่าวไม่ใช่คนเผือกเพราะไทโรซีนไม่เพียงเกิดขึ้นในร่างกายจากฟีนิลอะลานีนเท่านั้น แต่ยังเข้าสู่ร่างกายพร้อมกับอาหารอีกด้วย จริงอยู่ คนไข้ที่เป็นโรคฟีนิลคีโตนูเรียมักมีตาสีสว่าง ผิวขาว และมีผมสีขาว แน่นอนว่าอาจมีเผือกอยู่ด้วย แต่ถ้าบุคคลนั้นเป็นโฮโมไซกัสสำหรับยีนด้อยทั้งสองตัว
การถ่ายทอดทางพันธุกรรมของฟีนิลคีโตนูเรีย (PKU) อธิบายกฎของการแยก การกลายพันธุ์นี้เป็นแบบถอย เช่น สามารถต้านทานฟีโนไทป์ได้เฉพาะในสถานะโฮโมไซกัสเท่านั้น พบอุบัติการณ์สูงสุดของฟีนิลคีโตนูเรียในไอร์แลนด์ (16.4 รายต่อทารกแรกเกิด 100,000 คน); เพื่อการเปรียบเทียบ: ในสหรัฐอเมริกา - 5 รายต่อทารกแรกเกิด 100,000 คน
ยีน PKU และโครงสร้างที่แตกต่างกันซึ่งพบในประชากรที่แตกต่างกันได้รับการศึกษาอย่างดี ความรู้ที่มีอยู่ช่วยให้เราสามารถวินิจฉัยก่อนคลอดได้ทันท่วงทีเพื่อตรวจสอบว่าตัวอ่อนที่กำลังพัฒนาได้รับมรดกอัลลีล PKU สองสำเนาจากพ่อแม่ทั้งสองหรือไม่ (ข้อเท็จจริงของการถ่ายทอดทางพันธุกรรมดังกล่าวเพิ่มโอกาสของโรคอย่างรวดเร็ว) ในบางประเทศ เช่น อิตาลี ซึ่งมีอุบัติการณ์ของ PKU ค่อนข้างสูง การวินิจฉัยดังกล่าวเป็นสิ่งจำเป็นสำหรับหญิงตั้งครรภ์ทุกคน
PKU พบได้บ่อยมากในหมู่ผู้ที่แต่งงานกับญาติทางสายเลือด แม้ว่าอุบัติการณ์ของ PKU จะค่อนข้างต่ำ แต่ประมาณ 1 ใน 50 คนเป็นพาหะของ PKU allele ความน่าจะเป็นที่พาหะของอัลลีล PKU หนึ่งจะแต่งงานกับพาหะของอัลลีลอื่นคือประมาณ 2% อย่างไรก็ตาม เมื่อการแต่งงานเกิดขึ้นระหว่างญาติทางสายเลือด (เช่น หากคู่สมรสอยู่ในสายเลือดเดียวกันซึ่งได้รับอัลลีล PKU) ความเป็นไปได้ที่คู่สมรสทั้งสองจะเป็นพาหะของอัลลีล PKU และส่งต่ออัลลีลสองตัวไปสู่อนาคตพร้อมกัน เด็กจะสูงขึ้นอย่างมีนัยสำคัญ 2 %
ในกรณีของภาวะฟีนิลคีโตนูเรีย เรามีตัวอย่างที่ชัดเจนเกี่ยวกับวิธีการป้องกันการพัฒนาของโรคที่มีลักษณะทางพันธุกรรมโดยการเลือกอิทธิพลของสิ่งแวดล้อม ปัจจุบัน ฟีนิลคีโตนูเรียตรวจพบได้ง่ายในการตรวจทารกแรกเกิดเมื่ออายุ 2-3 วัน (โดยปกติความเข้มข้นของฟีนิลอะลานีนในพลาสมาไม่ควรเกิน 4 มก./ดล.) ผู้ป่วยจะได้รับฟีนิลอะลานีนในอาหารต่ำ ซึ่งจะช่วยหลีกเลี่ยงความเสียหายต่อพัฒนาการของระบบประสาท ในกรณีนี้ ไทโรซีนจะกลายเป็นกรดอะมิโนจำเป็น และจำเป็นเพื่อให้แน่ใจว่ามีอยู่ในอาหาร ช่วงเวลาที่สำคัญที่สุดคือระยะแรกของการสร้างเซลล์ ดังนั้นในวัยผู้ใหญ่ หลายคนไม่ปฏิบัติตามข้อ จำกัด ด้านอาหารอีกต่อไป แม้ว่าจะยังเป็นที่ต้องการก็ตาม ผู้หญิงที่เป็นโรคฟีนิลคีโตนูเรีย ไม่ว่าจะมีอาการใดก็ตาม จะต้องรับประทานอาหารพิเศษในระหว่างตั้งครรภ์ มิฉะนั้น ระดับฟีนิลอะลานีนในเลือดที่สูงจะส่งผลเสียหายต่อทารกในครรภ์ที่กำลังพัฒนา
Phenylketonuria เป็นตัวอย่างที่ดีของปฏิสัมพันธ์ระหว่างจีโนไทป์กับสิ่งแวดล้อม สาระสำคัญของโรคนี้คือความไวที่แตกต่างกันของบุคคลที่มีจีโนไทป์ต่างกันต่ออิทธิพลของสิ่งแวดล้อม สภาพแวดล้อมเดียวกัน (ในกรณีนี้สภาพแวดล้อมคือธรรมชาติของโภชนาการ) ทำให้เกิดการเจ็บป่วยที่รุนแรง (ฟีนิลคีโตนูเรีย) ในจีโนไทป์บางชนิด ในขณะที่จีโนไทป์อื่น ๆ ไม่มีการเปลี่ยนแปลงทางพยาธิวิทยาอย่างแน่นอน ภายใต้สภาพแวดล้อมอื่นๆ (ขึ้นอยู่กับอาหารพิเศษ) ความแตกต่างระหว่างจีโนไทป์สำหรับลักษณะนี้ (ฟีนิลคีโตนูเรีย) จะหายไป