O gráfico de linhagem mostra a herança da fenilcetonúria. Doenças hereditárias genéticas
A lei da clivagem também explica a herança da fenilcetonúria (PKU), uma doença que se desenvolve como resultado do excesso do importante aminoácido fenilalanina (Phe) no corpo humano. O excesso de fenilalanina leva ao desenvolvimento de retardo mental. A incidência de PKU é relativamente baixa (aproximadamente 1 em 10.000 nascimentos), no entanto, cerca de 1% dos indivíduos com atraso mental sofrem de PKU, constituindo assim um grupo relativamente grande de pacientes cujo atraso mental é explicado por um mecanismo genético homogéneo.
Tal como no caso do CG, os investigadores estudaram a incidência de PKU nas famílias dos probandos. Descobriu-se que os pacientes que sofrem de fenilcetonúria geralmente têm pais saudáveis. Além disso, observou-se que a FKU é mais comum em famílias cujos pais são parentes consanguíneos. Um exemplo de família de um probando que sofre de PKU é mostrado na Fig. 2.3: uma criança doente nasceu de pais fenotipicamente saudáveis que são parentes de sangue (primos), mas a irmã do pai da criança sofre de PKU.
Arroz. 2.3. Um exemplo de linhagem de uma família em que a PKU é herdada (a tia do probando sofre desta doença).
Uma linha dupla entre os cônjuges denota um casamento consanguíneo.
Os símbolos restantes são os mesmos da Fig. 2.1.
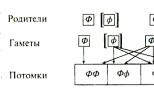
A PKU é transmitida por um modo de herança recessivo, ou seja, O genótipo do paciente contém dois alelos de PKU recebidos de ambos os pais. Os descendentes que possuem apenas um desses alelos não sofrem da doença, mas são portadores do alelo PKU” podem transmiti-la aos seus filhos. Na Fig. A Figura 2.4 mostra as formas de herança dos alelos da PKU de dois pais fenotipicamente normais. Cada pai tem um alelo PKU e um alelo normal. A probabilidade de cada filho herdar o alelo PKU de cada pai é de 50%. A probabilidade de uma criança herdar alelos de PKU de ambos os pais ao mesmo tempo é de 25% (0,5 x 0,5 = 0,25; as probabilidades são multiplicadas porque os eventos de herança de alelos de cada pai são independentes um do outro).
O gene PKU e suas variantes estruturais, encontradas em diferentes populações, têm sido bem estudadas. O conhecimento à nossa disposição permite-nos realizar um diagnóstico pré-natal oportuno para determinar se o feto em desenvolvimento herdou duas cópias do alelo PKU de ambos os pais (o facto de tal herança aumenta drasticamente a probabilidade da doença). Em alguns países, por exemplo na Itália, onde a incidência de PKU é bastante elevada, esse diagnóstico é obrigatório para todas as mulheres grávidas.

Arroz. 2.4. Esquema de cruzamento: mecanismo alélico de herança da PKU.
0 alelo dominante (“saudável”); [f] alelo recessivo que causa o desenvolvimento da doença. FF, FF são crianças fenotipicamente normais (75% delas): apenas 25% têm genótipo normal (FF); outros 50% são fenotipicamente saudáveis, mas são portadores do alelo PKU (Pf). Os 25% restantes dos descendentes estão doentes ([f][f])
Conforme observado, a PKU é mais comum entre aqueles que se casam com parentes consangüíneos. Embora a incidência de PKU seja relativamente baixa, aproximadamente 1 em cada 50 pessoas é portadora do alelo PKU. A probabilidade de um portador do alelo PKU se casar com outro portador desse alelo é de aproximadamente 2\%. No entanto, ao casar entre parentes consanguíneos (ou seja, se os cônjuges pertencem à mesma linhagem em que o alelo PKU é herdado), a probabilidade de ambos os cônjuges serem portadores do alelo PKU e transmitirem simultaneamente dois alelos ao nascituro será tornar-se significativamente maior 2\ %.
A lei da clivagem também explica a herança da fenilcetonúria
(PKU) - uma doença que se desenvolve como resultado do excesso de um importante
aminoácidos - fenilalanina (Phe) no corpo humano. Excesso
a fenilalanina leva ao desenvolvimento de retardo mental. Frequência
A incidência de PKU é relativamente baixa (aproximadamente 1 em 10.000 novos
nascido), no entanto, cerca de 1% dos indivíduos com retardo mental
mov sofrem de PKU, constituindo assim um grupo relativamente mais
maior grupo de pacientes cujo retardo mental é explicado
mecanismo genético homogêneo.
Assim como no caso do CG, os pesquisadores estudaram a frequência de ocorrência
PKU em famílias de probandos. Descobriu-se que os pacientes que sofrem de PKU
geralmente têm pais saudáveis. Além disso, percebeu-se que
A PKU é mais comum em famílias cujos pais são consangüíneos.
outros parentes. Exemplo de uma família de um probando que sofre de PKU

arroz. 2.3: doente
fenotípico
saudável
pais-
parentes
sofre
transmitido
herança,
doente
contém
recebido
pais.
Arroz. 2.3. Um exemplo de linhagem familiar, em
sofrer
transmitido
doença,
são
herança (a tia do probando sofre
detentores de alelos PKU e pode
esta doença).
entregar
Uma linha dupla entre os cônjuges significa
arroz. 2.4 mostrado
consanguíneo
Descansar
formação de alelos PKU de dois
as designações são as mesmas da Fig. 2.1.
fenotipicamente
normal
pais.
leu tem um alelo PKU e um alelo normal. Probabilidade
que toda criança pode herdar o alelo PKU de cada
dos pais é de 50%. A probabilidade de a criança ser
segue o alelo PKU de ambos os pais ao mesmo tempo, é de 25%
(0,5 x 0,5 = 0,25; as probabilidades são multiplicadas à medida que os eventos são herdados
os alelos de cada pai são independentes um do outro).
O gene PKU e suas variantes estruturais encontradas em diferentes
populações foram bem estudadas. O conhecimento à nossa disposição é
Arroz. 2.4. Esquema de cruzamento: mecanismo alélico de herança da PKU.
F - alelo dominante (“saudável”); [f] - alelo recessivo causador
desenvolvimento da doença. FF, FF - crianças fenotipicamente normais (75% delas); apenas
cerca de 25% têm genótipo normal (FF); outros 50% são fenotipicamente saudáveis,
mas são portadores do alelo PKU (FF). Os 25% restantes dos descendentes estão doentes
([f][f]).
casamento, permitir o diagnóstico pré-natal oportuno
tiques para determinar se o embrião em desenvolvimento herdou
respiram duas cópias do alelo PKU de ambos os pais (o fato de tal herança
vaniya aumenta drasticamente a probabilidade de doenças). Em alguns paises,
por exemplo, em Itália, onde a incidência de PKU é bastante elevada
suco, tais diagnósticos são realizados sem falhas para cada
ordenhar uma mulher grávida.
Como já foi observado, a PKU é mais comum entre aqueles que entram
casa com parentes de sangue. Apesar de a reunião
A incidência de PKU é relativamente baixa, aproximadamente 1 em cada 50 pessoas é
portador do alelo PKU. A probabilidade de que um portador do alelo
A PKU se casará com outro portador desse alelo, é
aproximadamente 2%. Porém, ao casar entre pessoas consanguíneas
parentes (ou seja, se os cônjuges pertencem à mesma linhagem, em
qual alelo PKU é herdado) a probabilidade de que
ambos os cônjuges serão portadores do alelo PKU e ao mesmo tempo transferirão
dará dois alelos ao feto, será significativamente superior a 2%.
 O método genealógico de estudo da hereditariedade é um dos métodos genéticos mais antigos e mais amplamente utilizados. A essência do método é compilar pedigrees que permitam traçar as características da herança dos traços. O método é aplicável se forem conhecidos os parentes diretos do proprietário da característica estudada nas linhas materna e paterna em várias gerações.
O método genealógico de estudo da hereditariedade é um dos métodos genéticos mais antigos e mais amplamente utilizados. A essência do método é compilar pedigrees que permitam traçar as características da herança dos traços. O método é aplicável se forem conhecidos os parentes diretos do proprietário da característica estudada nas linhas materna e paterna em várias gerações.

 Conteúdo 1. 2. 3. 4. 5. Símbolos Regras para elaboração de um pedigree Etapas da resolução de problemas Tipos de herança de características Resolução de problemas
Conteúdo 1. 2. 3. 4. 5. Símbolos Regras para elaboração de um pedigree Etapas da resolução de problemas Tipos de herança de características Resolução de problemas

 Regras para compilar pedigrees A pessoa de quem se começa a compilar um pedigree é chamada de probando. Os irmãos e irmãs do probando são chamados de irmãos. 1. O pedigree é representado de forma que cada geração esteja em sua própria linha horizontal. As gerações são numeradas com algarismos romanos e os membros da árvore genealógica são numerados com algarismos arábicos. 2. A elaboração do pedigree começa a partir do probando (dependendo do gênero - um quadrado ou círculo, indicado por uma seta) para que a partir dele seja possível traçar um pedigree tanto para baixo quanto para cima. 3. Ao lado do probando, coloque os símbolos de seus irmãos em ordem de nascimento (da esquerda para a direita), conectando-os com uma tecla gráfica.
Regras para compilar pedigrees A pessoa de quem se começa a compilar um pedigree é chamada de probando. Os irmãos e irmãs do probando são chamados de irmãos. 1. O pedigree é representado de forma que cada geração esteja em sua própria linha horizontal. As gerações são numeradas com algarismos romanos e os membros da árvore genealógica são numerados com algarismos arábicos. 2. A elaboração do pedigree começa a partir do probando (dependendo do gênero - um quadrado ou círculo, indicado por uma seta) para que a partir dele seja possível traçar um pedigree tanto para baixo quanto para cima. 3. Ao lado do probando, coloque os símbolos de seus irmãos em ordem de nascimento (da esquerda para a direita), conectando-os com uma tecla gráfica.
 4. Acima da linha do probando, indique os pais, conectando-os com a linha do casamento. 5. Na linha dos pais, desenhe os símbolos dos parentes mais próximos e dos seus cônjuges, ligando os seus graus de relacionamento em conformidade. 6. Na linha do probando, indique seus primos, etc., irmãos e irmãs, conectando-os de acordo com a linha dos pais. 7. Acima da linha dos pais, desenhe a linha dos avós. 8. Se o probando tiver filhos ou sobrinhos, coloque-os numa linha abaixo da linha do probando.
4. Acima da linha do probando, indique os pais, conectando-os com a linha do casamento. 5. Na linha dos pais, desenhe os símbolos dos parentes mais próximos e dos seus cônjuges, ligando os seus graus de relacionamento em conformidade. 6. Na linha do probando, indique seus primos, etc., irmãos e irmãs, conectando-os de acordo com a linha dos pais. 7. Acima da linha dos pais, desenhe a linha dos avós. 8. Se o probando tiver filhos ou sobrinhos, coloque-os numa linha abaixo da linha do probando.
 9. Após representar o pedigree (ou simultaneamente com ele), mostre apropriadamente os proprietários ou portadores heterozigotos da característica (na maioria das vezes, os portadores heterozigotos são determinados após a compilação e análise do pedigree). 10. Indique (se possível) os genótipos de todos os membros do pedigree. 11. Se houver várias doenças hereditárias na família que não estejam relacionadas entre si, crie um pedigree para cada doença separadamente.
9. Após representar o pedigree (ou simultaneamente com ele), mostre apropriadamente os proprietários ou portadores heterozigotos da característica (na maioria das vezes, os portadores heterozigotos são determinados após a compilação e análise do pedigree). 10. Indique (se possível) os genótipos de todos os membros do pedigree. 11. Se houver várias doenças hereditárias na família que não estejam relacionadas entre si, crie um pedigree para cada doença separadamente.
 Etapas da resolução do problema 1. Determine o tipo de herança do traço - dominante ou recessivo. Para isso, descubra: 1) se a característica em estudo é comum (em todas as gerações ou não); 2) quantos membros do pedigree possuem a característica; 3) se há casos de nascimento de filhos portadores do traço, caso os pais não apresentem esse traço; 4) se há casos de nascimento de filhos sem o traço estudado, se ambos os pais o possuírem; 5) que parte da prole carrega a característica nas famílias se um dos pais for seu dono.
Etapas da resolução do problema 1. Determine o tipo de herança do traço - dominante ou recessivo. Para isso, descubra: 1) se a característica em estudo é comum (em todas as gerações ou não); 2) quantos membros do pedigree possuem a característica; 3) se há casos de nascimento de filhos portadores do traço, caso os pais não apresentem esse traço; 4) se há casos de nascimento de filhos sem o traço estudado, se ambos os pais o possuírem; 5) que parte da prole carrega a característica nas famílias se um dos pais for seu dono.
 Etapas da resolução de problemas 2. Determine se a característica é herdada de maneira ligada ao sexo. Para isso, descubra: 1) com que frequência o sintoma ocorre em pessoas de ambos os sexos; se for raro, então qual gênero o carrega com mais frequência; 2) pessoas cujo gênero herdam a característica do pai e da mãe que carregam a característica.
Etapas da resolução de problemas 2. Determine se a característica é herdada de maneira ligada ao sexo. Para isso, descubra: 1) com que frequência o sintoma ocorre em pessoas de ambos os sexos; se for raro, então qual gênero o carrega com mais frequência; 2) pessoas cujo gênero herdam a característica do pai e da mãe que carregam a característica.
 Etapas da resolução do problema 3. Com base nos resultados da análise, tente determinar os genótipos de todos os membros do pedigree. Para determinar os genótipos, antes de mais nada, descubra a fórmula de divisão dos descendentes em uma geração.
Etapas da resolução do problema 3. Com base nos resultados da análise, tente determinar os genótipos de todos os membros do pedigree. Para determinar os genótipos, antes de mais nada, descubra a fórmula de divisão dos descendentes em uma geração.
 Tipos de herança de uma característica. 1. Herança autossômica dominante: 1) o traço ocorre com frequência no pedigree, em quase todas as gerações, com igual frequência em meninos e meninas; 2) se um dos pais for portador de uma característica, então essa característica aparecerá em todos os descendentes ou na metade.
Tipos de herança de uma característica. 1. Herança autossômica dominante: 1) o traço ocorre com frequência no pedigree, em quase todas as gerações, com igual frequência em meninos e meninas; 2) se um dos pais for portador de uma característica, então essa característica aparecerá em todos os descendentes ou na metade.
 O glaucoma é uma doença ocular caracterizada pelo aumento da pressão intraocular e diminuição da acuidade visual. Os fatores de risco para o desenvolvimento do glaucoma são: hereditariedade, diabetes mellitus, aterosclerose, trauma ocular, doenças oculares inflamatórias e degenerativas. Com a pressão intraocular constantemente elevada, a atrofia do nervo óptico se desenvolve gradualmente e a pessoa perde a visão. Braquidactilia (braquidactilia; braqui- + dedo grego daktylos; sinônimo de dedos curtos) é uma anomalia de desenvolvimento: encurtamento dos dedos das mãos ou dos pés. herdada de maneira autossômica dominante.
O glaucoma é uma doença ocular caracterizada pelo aumento da pressão intraocular e diminuição da acuidade visual. Os fatores de risco para o desenvolvimento do glaucoma são: hereditariedade, diabetes mellitus, aterosclerose, trauma ocular, doenças oculares inflamatórias e degenerativas. Com a pressão intraocular constantemente elevada, a atrofia do nervo óptico se desenvolve gradualmente e a pessoa perde a visão. Braquidactilia (braquidactilia; braqui- + dedo grego daktylos; sinônimo de dedos curtos) é uma anomalia de desenvolvimento: encurtamento dos dedos das mãos ou dos pés. herdada de maneira autossômica dominante.
 Tipos de herança de uma característica. 2. Herança autossômica recessiva: 1) o traço é raro, não em todas as gerações, sendo igualmente comum em meninos e meninas; 2) o traço pode aparecer nos filhos, mesmo que os pais não possuam esse traço; 3) se um dos pais for portador do traço, então ele não aparecerá nos filhos ou aparecerá na metade dos descendentes.
Tipos de herança de uma característica. 2. Herança autossômica recessiva: 1) o traço é raro, não em todas as gerações, sendo igualmente comum em meninos e meninas; 2) o traço pode aparecer nos filhos, mesmo que os pais não possuam esse traço; 3) se um dos pais for portador do traço, então ele não aparecerá nos filhos ou aparecerá na metade dos descendentes.
 O que é fenilcetonúria? A fenilcetonúria (PKU) é uma doença hereditária que aumenta a quantidade do aminoácido fenilalanina no sangue a níveis prejudiciais. (Os aminoácidos são os blocos de construção das proteínas). Se a PKU não for tratada, o excesso de fenilalanina pode causar retardo mental e outros problemas graves de saúde. Como as pessoas herdam PKU? A PKU é herdada de forma autossômica recessiva, o que significa que duas cópias do gene devem ser alteradas para que uma pessoa seja afetada pela doença. Na maioria das vezes, os pais de uma criança com doença autossômica recessiva não são afetados, mas são portadores de uma cópia do gene alterado.
O que é fenilcetonúria? A fenilcetonúria (PKU) é uma doença hereditária que aumenta a quantidade do aminoácido fenilalanina no sangue a níveis prejudiciais. (Os aminoácidos são os blocos de construção das proteínas). Se a PKU não for tratada, o excesso de fenilalanina pode causar retardo mental e outros problemas graves de saúde. Como as pessoas herdam PKU? A PKU é herdada de forma autossômica recessiva, o que significa que duas cópias do gene devem ser alteradas para que uma pessoa seja afetada pela doença. Na maioria das vezes, os pais de uma criança com doença autossômica recessiva não são afetados, mas são portadores de uma cópia do gene alterado.
 Tipos de herança de uma característica. 3. Herança ligada ao sexo: 1) X - herança dominante: ü o traço é mais comum no sexo feminino; ü se a mãe está doente e o pai saudável, a característica é transmitida aos filhos independentemente do sexo, podendo se manifestar tanto em meninas quanto em meninos; ü se a mãe estiver saudável e o pai doente, todas as filhas apresentarão o sintoma, mas os filhos não.
Tipos de herança de uma característica. 3. Herança ligada ao sexo: 1) X - herança dominante: ü o traço é mais comum no sexo feminino; ü se a mãe está doente e o pai saudável, a característica é transmitida aos filhos independentemente do sexo, podendo se manifestar tanto em meninas quanto em meninos; ü se a mãe estiver saudável e o pai doente, todas as filhas apresentarão o sintoma, mas os filhos não.
 3. Herança ligada ao sexo: 2) X - herança recessiva: o traço é mais encontrado no sexo masculino; Mais frequentemente, o sintoma se manifesta após uma geração; Se ambos os pais são saudáveis, mas a mãe é heterozigota, então a característica aparece frequentemente em 50% dos filhos; Se o pai estiver doente e a mãe for heterozigota, as mulheres também podem ter o traço.
3. Herança ligada ao sexo: 2) X - herança recessiva: o traço é mais encontrado no sexo masculino; Mais frequentemente, o sintoma se manifesta após uma geração; Se ambos os pais são saudáveis, mas a mãe é heterozigota, então a característica aparece frequentemente em 50% dos filhos; Se o pai estiver doente e a mãe for heterozigota, as mulheres também podem ter o traço.

 3. Herança ligada ao sexo: 3) Herança ligada ao Y: o traço ocorre apenas em homens; Se o pai carrega uma característica, então, via de regra, todos os filhos também possuem essa característica.
3. Herança ligada ao sexo: 3) Herança ligada ao Y: o traço ocorre apenas em homens; Se o pai carrega uma característica, então, via de regra, todos os filhos também possuem essa característica.
 Um exemplo de solução do problema O probando é uma mulher destra. Suas duas irmãs são destras, seus dois irmãos são canhotos. A mãe é destra. Ela tem dois irmãos e uma irmã, todos destros. Avó e avô são destros. O pai do probando é canhoto, sua irmã e seu irmão são canhotos, os outros dois irmãos e irmã são destros. Solução: 1. Desenhe o símbolo do probando. Mostramos a presença do sinal no probando.
Um exemplo de solução do problema O probando é uma mulher destra. Suas duas irmãs são destras, seus dois irmãos são canhotos. A mãe é destra. Ela tem dois irmãos e uma irmã, todos destros. Avó e avô são destros. O pai do probando é canhoto, sua irmã e seu irmão são canhotos, os outros dois irmãos e irmã são destros. Solução: 1. Desenhe o símbolo do probando. Mostramos a presença do sinal no probando.
 2. Colocamos os símbolos de seus irmãos ao lado do símbolo do probando. Nós os conectamos com um rocker gráfico.
2. Colocamos os símbolos de seus irmãos ao lado do símbolo do probando. Nós os conectamos com um rocker gráfico.




 7. Determine os genótipos dos membros do pedigree. O sinal de destro aparece em todas as gerações, tanto em mulheres quanto em homens. Isso indica um tipo de herança autossômica dominante da característica. I A- A- II A- A- A- Aa aa A- III aa Aa Aa A- aa
7. Determine os genótipos dos membros do pedigree. O sinal de destro aparece em todas as gerações, tanto em mulheres quanto em homens. Isso indica um tipo de herança autossômica dominante da característica. I A- A- II A- A- A- Aa aa A- III aa Aa Aa A- aa
 Tarefa 2. Com base no pedigree mostrado na figura, determine a natureza da manifestação do traço indicado em preto (dominante, recessivo, ligado ao sexo ou não). Determine o genótipo dos pais e filhos da primeira geração.
Tarefa 2. Com base no pedigree mostrado na figura, determine a natureza da manifestação do traço indicado em preto (dominante, recessivo, ligado ao sexo ou não). Determine o genótipo dos pais e filhos da primeira geração.
 Esquema de resolução do problema: 1) O traço recessivo não está ligado ao sexo; 2) Genótipos dos pais: mãe - aa, pai - AA ou Aa 3) Genótipos dos filhos: filho e filha heterozigotos - Aa.
Esquema de resolução do problema: 1) O traço recessivo não está ligado ao sexo; 2) Genótipos dos pais: mãe - aa, pai - AA ou Aa 3) Genótipos dos filhos: filho e filha heterozigotos - Aa.
 Tarefa 3 Utilizando o pedigree mostrado no diagrama, estabeleça o tipo e a natureza da manifestação do traço destacado em preto (dominante, recessivo, ligado ao sexo ou não). Determine os genótipos das crianças da primeira geração.
Tarefa 3 Utilizando o pedigree mostrado no diagrama, estabeleça o tipo e a natureza da manifestação do traço destacado em preto (dominante, recessivo, ligado ao sexo ou não). Determine os genótipos das crianças da primeira geração.
 Esquema de resolução do problema: 1) O traço é recessivo, ligado ao cromossomo X; 2) Genótipos dos pais: mãe – XHA, pai – XAU; 3) Genótipos de crianças em F 1: filho - Ha. Uh, filha - HAHA filha - HAHA
Esquema de resolução do problema: 1) O traço é recessivo, ligado ao cromossomo X; 2) Genótipos dos pais: mãe – XHA, pai – XAU; 3) Genótipos de crianças em F 1: filho - Ha. Uh, filha - HAHA filha - HAHA
 Tarefa 4 Utilizando o pedigree da pessoa mostrado na figura, estabeleça a natureza da herança do traço “olhos pequenos”, destacado em preto (dominante ou recessivo, ligado ao sexo ou não). Determine os genótipos dos pais e descendentes F 1 (1, 2, 3, 4, 5). 1 2 3 4 5
Tarefa 4 Utilizando o pedigree da pessoa mostrado na figura, estabeleça a natureza da herança do traço “olhos pequenos”, destacado em preto (dominante ou recessivo, ligado ao sexo ou não). Determine os genótipos dos pais e descendentes F 1 (1, 2, 3, 4, 5). 1 2 3 4 5
 Esquema para resolver o problema: 1) O traço é recessivo, não ligado ao sexo; 2) Genótipos dos pais: mãe – Aa, pai – Aa; 3) Genótipos dos descendentes em F 1: 1, 2 – Aa, 3, 5 – AA ou Aa; 4 – aa.
Esquema para resolver o problema: 1) O traço é recessivo, não ligado ao sexo; 2) Genótipos dos pais: mãe – Aa, pai – Aa; 3) Genótipos dos descendentes em F 1: 1, 2 – Aa, 3, 5 – AA ou Aa; 4 – aa.
 Codificador de elementos de conteúdo em biologia 3. 4 Genética, suas tarefas. Hereditariedade e variabilidade são propriedades dos organismos. Métodos genéticos. Conceitos genéticos básicos e simbolismo. Teoria cromossômica da hereditariedade. Idéias modernas sobre o gene e o genoma. 3. 5 Padrões de hereditariedade, sua base citológica. Padrões de herança estabelecidos por G. Mendel, sua base citológica (cruzamento mono e diíbrido). Leis de Morgan: herança vinculada de características, ruptura da ligação genética. Genética do sexo. Herança de características ligadas ao sexo. Interação genética. Genótipo como sistema integral. Genética humana. Métodos para estudar a genética humana. Resolvendo problemas genéticos. Elaboração de esquemas de travessia.
Codificador de elementos de conteúdo em biologia 3. 4 Genética, suas tarefas. Hereditariedade e variabilidade são propriedades dos organismos. Métodos genéticos. Conceitos genéticos básicos e simbolismo. Teoria cromossômica da hereditariedade. Idéias modernas sobre o gene e o genoma. 3. 5 Padrões de hereditariedade, sua base citológica. Padrões de herança estabelecidos por G. Mendel, sua base citológica (cruzamento mono e diíbrido). Leis de Morgan: herança vinculada de características, ruptura da ligação genética. Genética do sexo. Herança de características ligadas ao sexo. Interação genética. Genótipo como sistema integral. Genética humana. Métodos para estudar a genética humana. Resolvendo problemas genéticos. Elaboração de esquemas de travessia.
 ESPECIFICAÇÃO da prova de biologia A 7. Genética, suas tarefas, conceitos genéticos básicos. A 8. Padrões de hereditariedade. Genética humana. A 9. Padrões de variabilidade. A 30. Padrões genéticos. A influência dos mutagênicos no aparato genético de células e organismos. C 6. Resolver problemas de genética para aplicar o conhecimento em uma nova situação.
ESPECIFICAÇÃO da prova de biologia A 7. Genética, suas tarefas, conceitos genéticos básicos. A 8. Padrões de hereditariedade. Genética humana. A 9. Padrões de variabilidade. A 30. Padrões genéticos. A influência dos mutagênicos no aparato genético de células e organismos. C 6. Resolver problemas de genética para aplicar o conhecimento em uma nova situação.
 Parte A 1. A genética é de grande importância para a medicina, pois 1) combate epidemias 2) cria medicamentos para tratar pacientes 3) estabelece as causas das doenças hereditárias 4) protege o meio ambiente da poluição por mutagênicos
Parte A 1. A genética é de grande importância para a medicina, pois 1) combate epidemias 2) cria medicamentos para tratar pacientes 3) estabelece as causas das doenças hereditárias 4) protege o meio ambiente da poluição por mutagênicos
 2. O método usado para estudar a natureza da manifestação de características em irmãs ou irmãos que se desenvolveram a partir de um óvulo fertilizado é denominado 1. 2. 3. 4. Gêmeo Citogenético Genealógico Hibridológico
2. O método usado para estudar a natureza da manifestação de características em irmãs ou irmãos que se desenvolveram a partir de um óvulo fertilizado é denominado 1. 2. 3. 4. Gêmeo Citogenético Genealógico Hibridológico
 3. O método genealógico é usado para 1) obtenção de mutações genéticas e genômicas 2) estudo da influência da educação na ontogênese humana 3) estudo de doenças humanas hereditárias 4) estudo dos estágios de evolução do mundo orgânico
3. O método genealógico é usado para 1) obtenção de mutações genéticas e genômicas 2) estudo da influência da educação na ontogênese humana 3) estudo de doenças humanas hereditárias 4) estudo dos estágios de evolução do mundo orgânico
 4. Qual a função das consultas médicas de genética para casais parentais? 1. Identifica a predisposição dos pais para doenças infecciosas 2. Determina a possibilidade de ter gêmeos 3. Determina a probabilidade de doenças hereditárias nos filhos 4. Identifica a predisposição dos pais para distúrbios metabólicos
4. Qual a função das consultas médicas de genética para casais parentais? 1. Identifica a predisposição dos pais para doenças infecciosas 2. Determina a possibilidade de ter gêmeos 3. Determina a probabilidade de doenças hereditárias nos filhos 4. Identifica a predisposição dos pais para distúrbios metabólicos
 Determinar o genótipo pelo fenótipo A cor dos olhos de uma pessoa é determinada por um gene autossômico; o daltonismo é um gene recessivo ligado ao sexo. Determine o genótipo de uma mulher de olhos castanhos com visão normal de cores, cujo pai é daltônico (os olhos castanhos dominam os olhos azuis) 1) AAXDXD 3) Aa. Xd 2) Aa. XDXd 4) aa. XDXd
Determinar o genótipo pelo fenótipo A cor dos olhos de uma pessoa é determinada por um gene autossômico; o daltonismo é um gene recessivo ligado ao sexo. Determine o genótipo de uma mulher de olhos castanhos com visão normal de cores, cujo pai é daltônico (os olhos castanhos dominam os olhos azuis) 1) AAXDXD 3) Aa. Xd 2) Aa. XDXd 4) aa. XDXd
 Parte C Solução de problemas genéticos sobre a aplicação de conhecimentos em uma nova situação: cruzamento diíbrido, herança de características ligadas ao sexo, herança ligada de características (com cruzamento, sem cruzamento), determinação de grupos sanguíneos, análise de pedigree
Parte C Solução de problemas genéticos sobre a aplicação de conhecimentos em uma nova situação: cruzamento diíbrido, herança de características ligadas ao sexo, herança ligada de características (com cruzamento, sem cruzamento), determinação de grupos sanguíneos, análise de pedigree
 Parte C Em humanos, a herança do albinismo não está ligada ao sexo (A - a presença de melanina nas células da pele, e - a ausência de melanina nas células da pele - albinismo), e a hemofilia está ligada ao sexo (XH - coagulação sanguínea normal , Xh - hemofilia). Determinar os genótipos dos pais, bem como os possíveis genótipos, sexo e fenótipos dos filhos provenientes do casamento de uma mulher dihomozigota, normal para ambos os alelos, e de um homem albino com hemofilia. Faça um diagrama para resolver o problema.
Parte C Em humanos, a herança do albinismo não está ligada ao sexo (A - a presença de melanina nas células da pele, e - a ausência de melanina nas células da pele - albinismo), e a hemofilia está ligada ao sexo (XH - coagulação sanguínea normal , Xh - hemofilia). Determinar os genótipos dos pais, bem como os possíveis genótipos, sexo e fenótipos dos filhos provenientes do casamento de uma mulher dihomozigota, normal para ambos os alelos, e de um homem albino com hemofilia. Faça um diagrama para resolver o problema.
 O esquema para resolução do problema inclui: 1) genótipos dos pais: ♀AAXHXH (gametas AXH); ♂aa. X. Y (gametas a. Xh, a. Y); 2) genótipos e gênero das crianças: ♀Aa. XHXh; ♂Ah. XHY; 3) fenótipos de crianças: uma menina aparentemente normal para ambos os alelos, mas portadora dos genes do albinismo e da hemofilia; Um menino aparentemente normal para ambos os alelos, mas portador do gene do albinismo.
O esquema para resolução do problema inclui: 1) genótipos dos pais: ♀AAXHXH (gametas AXH); ♂aa. X. Y (gametas a. Xh, a. Y); 2) genótipos e gênero das crianças: ♀Aa. XHXh; ♂Ah. XHY; 3) fenótipos de crianças: uma menina aparentemente normal para ambos os alelos, mas portadora dos genes do albinismo e da hemofilia; Um menino aparentemente normal para ambos os alelos, mas portador do gene do albinismo.
Anomalias que levam ao aumento dos níveis fenilalanina sangue, na maioria das vezes deficiência de fenilalanina hidroxilase (PAH) ou fenilcetonúria (PKU), ilustram quase todos os princípios da genética bioquímica relacionados a defeitos enzimáticos. Todas as anomalias genéticas do metabolismo da fenilalanina são o resultado de mutações de perda de função no gene que codifica a HAP ou nos genes necessários para a síntese ou restauração do seu cofator, BH4.
Fenilcetonúria clássica(PKU) é legitimamente considerada um representante exemplar dos erros inatos do metabolismo. É um distúrbio autossômico recessivo da degradação da fenilalanina causado por mutações no gene que codifica a PAH, a enzima que converte a fenilalanina em tirosina. A descoberta da fenilcetonúria (PKU) por Fehling em 1934 foi a primeira a demonstrar um defeito genético como causa de retardo mental.
Devido à incapacidade de reciclar fenilalanina pacientes com fenilcetonúria (PKU) acumulam esse aminoácido nos fluidos corporais. A hiperfenilalaninemia danifica o sistema nervoso central em desenvolvimento na primeira infância e interfere no funcionamento do cérebro maduro. Uma pequena porção da fenilalanina é metabolizada por vias alternativas, produzindo quantidades aumentadas de ácido fenilpirúvico (o cetoácido que dá nome à doença) e outros metabólitos excretados na urina.
É interessante que embora defeito enzimáticoé conhecido há décadas, o mecanismo patogenético exato de como o aumento da fenilalanina danifica o cérebro ainda é desconhecido. É importante ressaltar que o desenvolvimento de danos neurológicos causados pelo bloqueio metabólico na PKU clássica pode ser amplamente prevenido por mudanças na dieta que evitem o acúmulo de fenilalanina. O tratamento da fenilcetonúria (PKU) tornou-se um modelo para o tratamento de muitas doenças metabólicas, cujos resultados podem ser melhorados através da prevenção da acumulação de substrato enzimático e seus derivados.
Triagem neonatal para fenilcetonúria (PKU)
A população é amplamente utilizada triagem recém-nascidos para fenilcetonúria (PKU). A fenilcetonúria (PKU) é um exemplo de doenças genéticas para as quais se justifica o rastreio neonatal em massa; a doença é relativamente comum em diversas populações (até 1 em 2.900 recém-nascidos vivos). O tratamento iniciado cedo na vida é muito eficaz; sem tratamento, inevitavelmente se desenvolve retardo mental grave. Os testes de triagem são realizados alguns dias após o nascimento.
Uma gota de sangue obtida de uma punção salto, aplicado em papel filtro, seco e enviado a um laboratório centralizado para avaliação dos níveis sanguíneos de fenilalanina e da relação fenilalanina/tirosina. No passado, as amostras eram coletadas antes do bebê receber alta hospitalar. A tendência de alta precoce da mãe e do recém-nascido após o parto alterou esta prática. É preferível não fazer o teste antes das 24 horas de idade porque os níveis de fenilalanina na fenilcetonúria (PKU) só aumentam depois do nascimento. Os resultados positivos dos testes devem ser rapidamente confirmados, uma vez que atrasar o início do tratamento mais de 4 semanas após o parto não evita o impacto no estado intelectual de pacientes com fenilcetonúria (PKU).
Várias formas de fenilcetonúria e hiperfenilalaninemia
Como a (PKU) está associada a uma deficiência grave da atividade da fenilalanina hidroxilase (PAH) (menos de 1% em comparação com os controles), a PAH mutante com atividade residual causa manifestações fenotípicas menos graves, a chamada hiperfenilalaninemia e fenilcetonúria atípica (PKU).
Hiperfenilalaninemia a fenilcetonúria (PKU), diferente da fenilcetonúria (PKU), é diagnosticada se a concentração plasmática de fenilalanina estiver abaixo de 1 mmol/L na presença de uma dieta normal. Este grau de hiperfenilalaninemia é apenas 10 vezes maior que o normal e significativamente menor que as concentrações encontradas na fenilcetonúria clássica (PKU) (>1 mmol/L). Não é provável que um aumento moderado de fenilalanina na hiperfenilalaninemia prejudique a função cerebral e pode até ser benéfico se o aumento for pequeno (<0,4 ммоль), такие дети обращают на себя внимание врачей только благодаря скринингу. Их нормальный фенотип оказался наилучшим показателем безопасного уровня фенилаланина плазмы, который не следует превышать при лечении пациентов с классической фенилкетонурии (ФКУ).
Atípico(PKU) - categoria que inclui pacientes com níveis de fenilalanina intermediários entre a PKU clássica e a hiperfenilalaninemia; esses pacientes necessitam de alguma restrição de fenilalanina na dieta, mas menos do que pacientes com fenilcetonúria clássica (PKU). O complexo destes três fenótipos clínicos com mutações no gene PAH é um exemplo de heterogeneidade clínica.
Hiperfenilalaninemia: heterogeneidade alélica e de locus na fenilcetonúria (PKU)
Molecular defeitos no gene da fenilalanina hidroxilase. Pacientes com hiperfenilalaninemia, incluindo fenilcetonúria clássica (PKU), fenilcetonúria atípica (PKU) e hiperfenilalaninemia benigna, apresentam um grau impressionante de heterogeneidade alélica no locus da fenilalanina hidroxilase (PAH) (mais de 400 mutações diferentes em todo o mundo).
A grande maioria dos alelos fenilalanina hidroxilase(PAH) são mutações bastante raras que perturbam as propriedades enzimáticas da fenilalanina hidroxilase (PAH) e levam à hiperfenilalaninemia, embora também tenham sido encontrados polimorfismos benignos ou variantes benignas menos comuns.
Em populações Descendência europeia cerca de dois terços dos cromossomos mutantes conhecidos são representados por seis mutações. Seis outras mutações são responsáveis por pouco mais de 80% das mutações da fenilalanina hidroxilase (PAH) nas populações asiáticas. Outras mutações patogênicas são menos comuns. Para tornar esta informação amplamente disponível, um consórcio internacional desenvolveu uma base de dados de mutações no gene da fenilalanina hidroxilase (PAH).
Em tudo populações Existe uma marcada heterogeneidade genética da fenilalanina hidroxilase (PAH). Devido ao alto grau de heterogeneidade alélica no locus, a maioria dos pacientes com fenilcetonúria (PKU) em muitas populações são heterozigotos compostos (isto é, eles têm dois alelos patogênicos diferentes), o que é totalmente consistente com a heterogeneidade enzimática e fenotípica observada em distúrbios da fenilalanina hidroxilase (PAH).

A princípio parecia que o conhecimento do genótipo fenilalanina hidroxilase(FA) prevê com segurança detalhes do fenótipo; essa expectativa não foi totalmente justificada, embora tenha sido encontrada certa correlação entre o genótipo da HAP e o fenótipo bioquímico.
Em termos gerais, mutações que suprimem completamente ou reduzem drasticamente a atividade fenilalanina hidroxilase(PAH) causam fenilcetonúria clássica (PKU), enquanto mutações que resultam em atividade enzimática residual suficientemente grande estão associadas a fenótipos leves.
No entanto, algumas mutações fenilalanina hidroxilase(FA) em pacientes homozigotos determinam todo o espectro de fenótipos, desde a fenilcetonúria clássica (PKU) até a hiperfenilalaninemia benigna.
Assim, ficou óbvio que na formação fenótipo observado em um genótipo específico, outros fatores biológicos não identificados estão envolvidos, incluindo, sem dúvida, genes modificadores. Esta observação, agora reconhecida como uma característica comum de muitas doenças monogénicas, indica que mesmo doenças monogénicas como a fenilcetonúria (PKU) não são doenças geneticamente simples.
Defeitos no metabolismo da tetrahidrobiopterina na fenilcetonúria (PKU)
Inicialmente acreditava-se que todas as crianças com doenças hereditárias hiperfenilalaninemia tem deficiência primária de fenilalanina hidroxilase (PAH). Está agora claro que aproximadamente 1-3% dos pacientes têm um gene normal da HAP e a sua hiperfenilalaninemia é o resultado de um defeito genético num dos vários outros genes envolvidos na síntese ou regeneração do cofator da HAP, BH4. A associação de um fenótipo, como a hiperfenilalaninemia, com mutações em genes diferentes é um exemplo de heterogeneidade de locus.
Como mostrado por mutações em genes codificadores de proteínas fenilalanina hidroxilase(PAH) e o metabolismo de seu cofator biopterina, proteínas codificadas por genes que exibem heterogeneidade de locus, geralmente participam da mesma cadeia de reações bioquímicas. Os pacientes com deficiência de BH4 foram identificados pela primeira vez porque, apesar de manterem com sucesso baixas concentrações de fenilalanina na dieta, desenvolveram problemas neurológicos profundos de início precoce.
Os maus resultados são parcialmente explicados a necessidade de cofator BH4 para a atividade de duas outras enzimas, tirosina hidroxilase e triptofano hidroxilase. Ambas as hidroxilases são críticas para a síntese de neurotransmissores monoamina, como dehidroxifenilalanina, norepinefrina, epinefrina e serotonina. Pacientes com deficiência de BH4 apresentam comprometimento na biossíntese do GTP ou na regeneração de BH4. Assim como a fenilcetonúria clássica (PKU), o distúrbio é herdado de maneira autossômica recessiva.
É importante distinguir pacientes com defeitos no metabolismo da BH4 de pacientes com mutações no fenilalanina hidroxilase(FA), uma vez que seu tratamento difere acentuadamente. Primeiro, uma vez que a estrutura proteica da fenilalanina hidroxilase (PAH) é normal em pacientes com distúrbios BH4, a sua atividade pode ser restaurada se estes pacientes receberem grandes doses de BH4, o que leva a uma diminuição nos níveis plasmáticos de fenilalanina. Portanto, o grau de restrição de fenilalanina na dieta de pacientes com defeitos no metabolismo da BH4 pode ser significativamente reduzido, e alguns pacientes podem ser mudados para uma dieta normal (isto é, sem restrição de fenilalanina).
Em segundo lugar, você também deve tentar normalizar níveis de neurotransmissores no cérebro desses pacientes pela administração de produtos tirosina hidroxilase e triptofano hidroxilase: L-dopa e 5-hidroxitriptofano, respectivamente. Por estas razões, todos os recém-nascidos com hiperfenilalaninemia devem ser avaliados quanto a anomalias no metabolismo da BH4.
Reação à tetrahidrobiopterina com mutações no gene PAH na fenilcetonúria (PKU)
Na maioria dos pacientes com mutações no gene fenilalanina hidroxilase(PAH), e não no metabolismo do BH4, houve uma clara diminuição do nível de fenilalanina no sangue durante a administração oral de grandes doses do cofator fenilalanina hidroxilase (PAH) BH4. Pacientes com atividade residual significativa da fenilalanina hidroxilase (PAH) (ou seja, pacientes com fenilcetonúria atípica (PKU) e hiperfenilalaninemia) respondem melhor a esse tratamento, mas um pequeno número de pacientes, mesmo com fenilcetonúria clássica (PKU), também responde a esse tratamento. Ao mesmo tempo, a presença de atividade residual de PAH não garante um efeito nos níveis plasmáticos de fenilalanina quando o BH4 é prescrito.
É mais provável que o grau de resposta reações na BH4 depende das propriedades específicas de cada proteína mutante da fenilalanina hidroxilase (PAH), refletindo a heterogeneidade alélica subjacente das mutações da PAH. Foi demonstrado que a introdução de BH4 na dieta tem efeito terapêutico através de diversos mecanismos causados pelo aumento da quantidade de cofator normal que entra em contato com o mutante.
Esses mecanismos incluem a estabilização do mutante enzima, proteção da enzima contra a degradação celular, aumento do fornecimento de um cofator para a enzima que tem baixa afinidade pela BH4 e outros efeitos benéficos nas propriedades cinéticas e catalíticas da enzima. Fornecer quantidades maiores de cofator é uma estratégia comum usada no tratamento de muitos erros inatos do metabolismo.
Uma série de mutações genéticas, nas quais a estrutura de apenas um gene é alterada, leva ao desenvolvimento de retardo mental. Segundo algumas estimativas, em 7 a 10% dos pacientes com oligofrenia ela é causada por mutações desse tipo.
O conjunto de reações bioquímicas que ocorrem no corpo é denominado metabolismo. Muitos genes codificam proteínas que participam como enzimas em certas reações metabólicas. Uma mutação em tal gene pode levar o corpo a produzir uma enzima menos ativa ou completamente inativa e, às vezes, à cessação completa da síntese enzimática. Nesse caso, a reação normalmente realizada por essa enzima fica mais lenta ou nem ocorre, o que causa o distúrbio hereditário correspondente - um dos chamados erros inatos do metabolismo. As doenças hereditárias genéticas mais comuns incluem fenilcetonúria, anemia falciforme, doença de Tay-Sachs, hemofilia e diabetes mellitus. A extensão em que influenciam o fenótipo depende da importância da enzima afetada para o organismo. Vimos acima que a doença de Tay-Sachs e a fibrose cística levam à morte. Algumas outras anomalias genéticas causam vários problemas graves no corpo, mas não são fatais.
A fenilcetonúria e o albinismo afetam a mesma via metabólica.
A fenilcetonúria é uma doença na qual, como resultado de uma mutação, a estrutura da enzima envolvida no metabolismo do aminoácido fenilalanina (fenilalanina hidroxilase) é perturbada. Esta enzima é necessária para a conversão da fenilalanina em tirosina. Doenças deste tipo são chamadas de enzimopatias, ou seja, causada por um defeito nas enzimas. Com esta doença, a fenilalanina e os produtos do seu metabolismo inadequado (ácido fenilacético) acumulam-se no sangue, o que causa danos ao sistema nervoso em desenvolvimento. Isto é principalmente a destruição da mielina e a degeneração do sistema nervoso espongiforme. Ocorrem retardo mental, microcefalia, psicose, tremor, atividade convulsiva e espasticidade.
A fenilcetonúria afeta indivíduos homozigotos para um gene recessivo que os priva da capacidade de sintetizar uma das enzimas necessárias para converter o aminoácido fenilalanina em outro aminoácido, a tirosina. Em vez de ser convertida em tirosina, a fenilalanina é convertida em ácido fenilpirúvico, que se acumula em quantidades tóxicas no sangue, afeta o cérebro e (se não for tratado prontamente) causa retardo mental. A urina dos pacientes também contém ácido fenilpirúvico, que lhe confere um odor característico. Atualmente, a fenilcetonúria é tratada com dieta especial. Para isso, nos primeiros anos de vida de uma criança, a fenilalanina é quase totalmente excluída de sua alimentação. Uma vez concluído o desenvolvimento do cérebro, uma paciente com fenilcetonúria é colocada em uma dieta normal, mas uma mulher com esse distúrbio genético deve seguir uma dieta pobre em fenilalanina durante a gravidez para prevenir o desenvolvimento anormal do cérebro fetal. Nos Estados Unidos, em muitos estados, todos os recém-nascidos são obrigados a submeter-se a testes especiais para PKU e alguns outros erros inatos do metabolismo.
Indivíduos homozigotos para o gene do albinismo não possuem a enzima que normalmente catalisa a conversão da tirosina em melanina, ou seja, pigmento que determina a cor marrom ou preta dos olhos, cabelos e pele. Os albinos têm cabelos brancos e pele e olhos muito claros. Naturalmente, pode surgir a questão de saber se os pacientes com fenilcetonúria também são albinos, uma vez que os seus corpos não produzem tirosina, a partir da qual a melanina é finalmente produzida. No entanto, esses pacientes não são albinos, porque a tirosina não é apenas formada no próprio corpo a partir da fenilalanina, mas também entra no corpo com os alimentos. É verdade que os pacientes com fenilcetonúria geralmente têm olhos claros, pele clara e cabelos louros. Claro, pode haver albinos entre eles, mas apenas se o indivíduo for homozigoto para ambos os genes recessivos.
A herança da fenilcetonúria (PKU) explica a lei da divisão. Esta mutação é recessiva, ou seja, pode resistir no fenótipo apenas no estado homozigoto. A maior incidência de fenilcetonúria foi observada na Irlanda (16,4 casos por 100 mil recém-nascidos); para efeito de comparação: nos EUA - 5 casos por 100 mil recém-nascidos.
O gene PKU e suas variantes estruturais, encontradas em diferentes populações, têm sido bem estudadas. O conhecimento à nossa disposição permite-nos realizar um diagnóstico pré-natal oportuno para determinar se o embrião em desenvolvimento herdou duas cópias do alelo PKU de ambos os pais (o facto de tal herança aumenta drasticamente a probabilidade da doença). Em alguns países, por exemplo na Itália, onde a incidência de PKU é bastante elevada, esse diagnóstico é obrigatório para todas as mulheres grávidas.
A PKU é mais comum entre aqueles que se casam com parentes consangüíneos. Embora a incidência de PKU seja relativamente baixa, aproximadamente 1 em cada 50 pessoas é portadora do alelo PKU. A probabilidade de um portador do alelo PKU se casar com outro portador desse alelo é de aproximadamente 2%. No entanto, quando ocorre um casamento entre parentes consanguíneos (ou seja, se os cônjuges pertencem à mesma linhagem em que o alelo PKU é herdado), a probabilidade de ambos os cônjuges serem portadores do alelo PKU e transmitirem simultaneamente dois alelos para o futuro criança será significativamente superior a 2%.
No caso da fenilcetonúria, temos um exemplo marcante de como o desenvolvimento de uma doença de natureza genética pode ser prevenido através da seleção de influências ambientais. Atualmente, a fenilcetonúria é facilmente detectada durante exames de rotina de recém-nascidos com 2 a 3 dias de idade (normalmente, a concentração de fenilalanina no plasma sanguíneo não deve exceder 4 mg/dL). Os pacientes são submetidos a uma dieta pobre em fenilalanina, o que ajuda a evitar danos ao desenvolvimento do sistema nervoso. Neste caso, a tirosina torna-se um aminoácido essencial e é necessário garantir a sua presença na dieta alimentar. O período mais crítico são os estágios iniciais da ontogênese, portanto, na idade adulta, muitos não aderem mais às restrições alimentares, embora isso ainda seja desejável. Mulheres com fenilcetonúria, independentemente da sua condição, devem seguir uma dieta especial durante a gravidez, caso contrário, os níveis elevados de fenilalanina no sangue terão um efeito prejudicial no feto em desenvolvimento.
A fenilcetonúria é um bom exemplo de interação genótipo-ambiente. A essência desta doença é a diferente sensibilidade de indivíduos com diferentes genótipos às influências ambientais. O mesmo ambiente (neste caso, o ambiente é a natureza da nutrição) causa uma doença grave (fenilcetonúria) em alguns genótipos, enquanto em outros genótipos não são observadas absolutamente nenhuma alteração patológica. Sob outras condições ambientais (sujeitas a uma dieta especial), as diferenças entre os genótipos para esta característica (fenilcetonúria) desaparecem.