Bagan silsilah menunjukkan pewarisan fenilketonuria. Penyakit keturunan genetik
Hukum pembelahan juga menjelaskan pewarisan fenilketonuria (PKU), suatu penyakit yang berkembang akibat kelebihan asam amino penting fenilalanin (Phe) dalam tubuh manusia. Kelebihan fenilalanin menyebabkan perkembangan keterbelakangan mental. Insiden PKU relatif rendah (sekitar 1 dari 10.000 kelahiran), namun sekitar 1% individu dengan keterbelakangan mental menderita PKU, sehingga merupakan kelompok pasien yang relatif besar yang keterbelakangan mentalnya disebabkan oleh mekanisme genetik yang homogen.
Seperti halnya CG, peneliti mempelajari kejadian PKU di keluarga pasien yang mengalami masalah. Ternyata pasien yang menderita PKU biasanya memiliki orang tua yang sehat. Selain itu, FKU telah diamati lebih sering terjadi pada keluarga yang orang tuanya adalah saudara sedarah. Contoh keluarga pasien yang menderita PKU ditunjukkan pada Gambar. 2.3: seorang anak yang sakit dilahirkan dari orang tua yang secara fenotip sehat merupakan saudara sedarah (sepupu), tetapi saudara perempuan dari ayah anak tersebut menderita PKU.
Beras. 2.3. Contoh silsilah keluarga yang mewarisi PKU (bibi calon menderita penyakit ini).
Garis ganda di antara pasangan menunjukkan perkawinan sedarah.
Simbol yang tersisa sama seperti pada Gambar. 2.1.
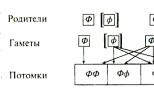
PKU ditularkan melalui pola pewarisan resesif, yaitu. Genotipe pasien mengandung dua alel PKU yang diperoleh dari kedua orang tuanya. Keturunan yang hanya memiliki satu alel tersebut tidak menderita penyakit tersebut, tetapi merupakan pembawa alel PKU” dapat menularkannya kepada anaknya. Pada Gambar. Gambar 2.4 menunjukkan cara pewarisan alel PKU dari dua orang tua yang berfenotip normal. Setiap orang tua mempunyai satu alel PKU dan satu alel normal. Peluang setiap anak mewarisi alel PKU dari masing-masing orang tuanya adalah 50%. Peluang seorang anak mewarisi alel PKU dari kedua orang tuanya pada saat yang sama adalah 25\% (0,5 x 0,5 = 0,25; peluangnya dikalikan karena kejadian pewarisan alel dari masing-masing orang tua tidak tergantung satu sama lain).
Gen PKU dan varian strukturalnya, yang ditemukan pada populasi berbeda, telah dipelajari dengan baik. Pengetahuan yang kami miliki memungkinkan kami melakukan diagnosis prenatal tepat waktu untuk menentukan apakah janin yang sedang berkembang mewarisi dua salinan alel PKU dari kedua orang tuanya (fakta pewarisan tersebut secara tajam meningkatkan kemungkinan penyakit). Di beberapa negara, misalnya di Italia, yang angka kejadian PKU cukup tinggi, diagnosis seperti itu wajib dilakukan oleh setiap ibu hamil.

Beras. 2.4. Skema persilangan: mekanisme pewarisan alelik PKU.
0 alel dominan (“sehat”); [f] alel resesif yang menyebabkan perkembangan penyakit. FF, FF adalah anak-anak yang fenotipnya normal (75% diantaranya): hanya 25% yang memiliki genotipe normal (FF); 50% lainnya memiliki fenotip yang sehat, namun merupakan pembawa alel PKU (Pf). Sisanya 25% keturunannya sakit ([f][f])
Sebagaimana telah disebutkan, PKU lebih sering terjadi pada mereka yang menikah dengan saudara sedarah. Meskipun kejadian PKU relatif rendah, sekitar 1 dari 50 orang adalah pembawa alel PKU. Probabilitas bahwa salah satu pembawa alel PKU akan menikah dengan pembawa alel lain yang serupa adalah sekitar 2\%. Namun, ketika menikah antara saudara sedarah (yaitu, jika pasangan memiliki silsilah yang sama di mana alel PKU diwariskan), kemungkinan besar kedua pasangan akan menjadi pembawa alel PKU dan secara bersamaan mewariskan dua alel kepada anak yang belum lahir. menjadi jauh lebih tinggi 2\ %.
Hukum pembelahan juga menjelaskan pewarisan fenilketonuria
(PKU) - penyakit yang berkembang akibat kelebihan suatu hal penting
asam amino - fenilalanin (Phe) dalam tubuh manusia. Kelebihan
fenilalanin menyebabkan perkembangan keterbelakangan mental. Frekuensi
Insiden PKU relatif rendah (kira-kira 1 dari 10.000 kasus baru
lahir), namun, sekitar 1% dari individu yang mengalami keterbelakangan mental
mov menderita PKU, sehingga membuat relatif lebih banyak
kelompok pasien terbesar yang keterbelakangan mentalnya dapat dijelaskan
mekanisme genetik yang homogen.
Seperti halnya CG, peneliti mempelajari frekuensi kejadiannya
PKU dalam keluarga masalah. Ternyata pasien menderita PKU
biasanya memiliki orang tua yang sehat. Selain itu, hal itu juga diperhatikan
PKU lebih sering terjadi pada keluarga dengan orang tua sedarah
kerabat lainnya. Contoh keluarga pasien yang menderita PKU

beras. 2.3: sakit
fenotipik
sehat
orang tua-
kerabat
menderita
ditularkan
warisan,
sakit
mengandung
diterima
orang tua.
Beras. 2.3. Contoh silsilah keluarga, di
menderita
ditularkan
penyakit,
adalah
warisan (bibi calon menderita
pemegang alel PKU dan bisa
penyakit ini).
serahkan
Artinya garis ganda antar pasangan
beras. 2.4 ditampilkan
kerabat
Istirahat
pembentukan alel PKU dari dua
sebutannya sama seperti pada Gambar. 2.1.
secara fenotip
normal
orang tua.
leu memiliki satu alel PKU dan satu alel normal. Kemungkinan
bahwa setiap anak dapat mewarisi alel PKU dari setiap anak
orang tua adalah 50%. Kemungkinan anak tersebut adalah
mengikuti alel PKU dari kedua orang tuanya secara bersamaan, adalah 25%
(0,5 x 0,5 = 0,25; probabilitas dikalikan seiring dengan pewarisan peristiwa
alel dari masing-masing orang tua tidak bergantung satu sama lain).
Gen PKU dan varian strukturalnya ditemukan berbeda
populasi telah dipelajari dengan baik. Pengetahuan yang kita miliki adalah
Beras. 2.4. Skema persilangan: mekanisme pewarisan alelik PKU.
F - alel dominan (“sehat”); [f] - penyebab alel resesif
perkembangan penyakit. FF, FF - anak-anak yang fenotipnya normal (75% di antaranya); hanya
sekitar 25% memiliki genotipe normal (FF); 50% lainnya sehat secara fenotip,
tetapi merupakan pembawa alel PKU (FF). Sisanya, 25% keturunannya sakit
([f][f]).
pernikahan, memungkinkan diagnosis prenatal tepat waktu
tics untuk menentukan apakah embrio yang sedang berkembang memiliki warisan
menghirup dua salinan alel PKU dari kedua orang tuanya (fakta pewarisan tersebut
vaniya secara tajam meningkatkan kemungkinan penyakit). Di beberapa negara,
misalnya di Italia yang angka kejadian PKU cukup tinggi
jus, diagnosis semacam itu dilakukan tanpa gagal untuk semua orang
memerah susu wanita hamil.
Seperti yang telah disebutkan, PKU lebih sering terjadi pada mereka yang masuk
menikah dengan saudara sedarah. Terlepas dari kenyataan bahwa pertemuan itu
Angka kejadian PKU relatif rendah, sekitar 1 dari 50 orang
pembawa alel PKU. Peluang bahwa salah satu pembawa alel
PKU akan mengawini pembawa alel lain yang serupa
sekitar 2%. Namun bila menikah antar saudara kandung
kerabat (yaitu jika pasangan memiliki silsilah yang sama, di
alel PKU mana yang diwarisi) kemungkinannya
kedua pasangan akan menjadi pembawa alel PKU dan sekaligus berpindah
akan memberikan dua alel kepada anak yang belum lahir, maka akan menjadi jauh lebih tinggi dari 2%.
 Metode silsilah dalam mempelajari hereditas adalah salah satu metode genetika tertua dan paling banyak digunakan. Inti dari metode ini adalah menyusun silsilah yang memungkinkan seseorang menelusuri ciri-ciri pewarisan sifat. Metode ini dapat diterapkan jika diketahui kerabat langsung dari pemilik sifat yang diteliti melalui garis ibu dan ayah dalam beberapa generasi.
Metode silsilah dalam mempelajari hereditas adalah salah satu metode genetika tertua dan paling banyak digunakan. Inti dari metode ini adalah menyusun silsilah yang memungkinkan seseorang menelusuri ciri-ciri pewarisan sifat. Metode ini dapat diterapkan jika diketahui kerabat langsung dari pemilik sifat yang diteliti melalui garis ibu dan ayah dalam beberapa generasi.

 Daftar Isi 1. 2. 3. 4. 5. Simbol Aturan menyusun silsilah Tahapan pemecahan masalah Jenis pewarisan sifat Pemecahan masalah
Daftar Isi 1. 2. 3. 4. 5. Simbol Aturan menyusun silsilah Tahapan pemecahan masalah Jenis pewarisan sifat Pemecahan masalah

 Aturan Penyusunan Silsilah Orang dari siapa mereka mulai menyusun silsilah disebut proband. Saudara laki-laki dan perempuan proband disebut saudara kandung. 1. Silsilah digambarkan sedemikian rupa sehingga setiap generasi berada pada garis horizontalnya masing-masing. Generasi diberi nomor dengan angka Romawi, dan anggota silsilah keluarga diberi nomor dengan angka Arab. 2. Penyusunan silsilah dimulai dari proband (tergantung jenis kelamin - persegi atau lingkaran, ditandai dengan panah) sehingga darinya dimungkinkan untuk menggambar silsilah ke bawah dan ke atas. 3. Di sebelah proband, letakkan lambang saudara kandungnya sesuai urutan lahir (dari kiri ke kanan), hubungkan dengan grafis rocker.
Aturan Penyusunan Silsilah Orang dari siapa mereka mulai menyusun silsilah disebut proband. Saudara laki-laki dan perempuan proband disebut saudara kandung. 1. Silsilah digambarkan sedemikian rupa sehingga setiap generasi berada pada garis horizontalnya masing-masing. Generasi diberi nomor dengan angka Romawi, dan anggota silsilah keluarga diberi nomor dengan angka Arab. 2. Penyusunan silsilah dimulai dari proband (tergantung jenis kelamin - persegi atau lingkaran, ditandai dengan panah) sehingga darinya dimungkinkan untuk menggambar silsilah ke bawah dan ke atas. 3. Di sebelah proband, letakkan lambang saudara kandungnya sesuai urutan lahir (dari kiri ke kanan), hubungkan dengan grafis rocker.
 4. Di atas garis perpisahan, tunjukkan orang tua, yang menghubungkan mereka satu sama lain dengan garis perkawinan. 5. Pada garis orang tua, gambarlah lambang kerabat terdekat dan pasangannya, serta hubungkan derajat kekerabatan mereka. 6. Pada garis proband, sebutkan sepupu-sepupunya, dsb., saudara-saudaranya, dan hubungkan mereka sesuai dengan garis orangtuanya. 7. Di atas garis orang tua, gambarlah garis kakek dan nenek. 8. Jika proband mempunyai anak atau keponakan, tempatkan mereka pada garis di bawah garis proband.
4. Di atas garis perpisahan, tunjukkan orang tua, yang menghubungkan mereka satu sama lain dengan garis perkawinan. 5. Pada garis orang tua, gambarlah lambang kerabat terdekat dan pasangannya, serta hubungkan derajat kekerabatan mereka. 6. Pada garis proband, sebutkan sepupu-sepupunya, dsb., saudara-saudaranya, dan hubungkan mereka sesuai dengan garis orangtuanya. 7. Di atas garis orang tua, gambarlah garis kakek dan nenek. 8. Jika proband mempunyai anak atau keponakan, tempatkan mereka pada garis di bawah garis proband.
 9. Setelah menggambarkan silsilah (atau bersamaan dengan itu), tunjukkan dengan tepat pemilik atau pembawa sifat heterozigot (paling sering, pembawa heterozigot ditentukan setelah kompilasi dan analisis silsilah). 10. Tunjukkan (jika mungkin) genotipe semua anggota silsilah. 11. Jika dalam suatu keluarga terdapat beberapa penyakit keturunan yang tidak berkaitan satu sama lain, buatlah silsilah masing-masing penyakit secara terpisah.
9. Setelah menggambarkan silsilah (atau bersamaan dengan itu), tunjukkan dengan tepat pemilik atau pembawa sifat heterozigot (paling sering, pembawa heterozigot ditentukan setelah kompilasi dan analisis silsilah). 10. Tunjukkan (jika mungkin) genotipe semua anggota silsilah. 11. Jika dalam suatu keluarga terdapat beberapa penyakit keturunan yang tidak berkaitan satu sama lain, buatlah silsilah masing-masing penyakit secara terpisah.
 Tahapan pemecahan masalah 1. Menentukan jenis pewarisan sifat – dominan atau resesif. Untuk melakukannya, cari tahu: 1) apakah sifat yang dipelajari itu umum (di semua generasi atau tidak); 2) berapa banyak anggota silsilah yang memiliki sifat tersebut; 3) apakah ada kasus kelahiran anak yang memiliki sifat tersebut, jika orang tuanya tidak menunjukkan sifat tersebut; 4) apakah ada kasus kelahiran anak tanpa sifat yang diteliti, jika kedua orang tuanya memilikinya; 5) bagian manakah dari keturunan yang membawa sifat tersebut dalam keluarga jika salah satu orang tuanya adalah pemiliknya.
Tahapan pemecahan masalah 1. Menentukan jenis pewarisan sifat – dominan atau resesif. Untuk melakukannya, cari tahu: 1) apakah sifat yang dipelajari itu umum (di semua generasi atau tidak); 2) berapa banyak anggota silsilah yang memiliki sifat tersebut; 3) apakah ada kasus kelahiran anak yang memiliki sifat tersebut, jika orang tuanya tidak menunjukkan sifat tersebut; 4) apakah ada kasus kelahiran anak tanpa sifat yang diteliti, jika kedua orang tuanya memilikinya; 5) bagian manakah dari keturunan yang membawa sifat tersebut dalam keluarga jika salah satu orang tuanya adalah pemiliknya.
 Tahapan pemecahan masalah 2. Menentukan apakah sifat tersebut diturunkan secara terpaut seks. Untuk melakukannya, cari tahu: 1) seberapa sering gejala tersebut terjadi pada kedua jenis kelamin; jika jarang, jenis kelamin mana yang lebih sering mengidapnya; 2) orang yang berjenis kelamin mewarisi sifat tersebut dari ayah dan ibu yang membawa sifat tersebut.
Tahapan pemecahan masalah 2. Menentukan apakah sifat tersebut diturunkan secara terpaut seks. Untuk melakukannya, cari tahu: 1) seberapa sering gejala tersebut terjadi pada kedua jenis kelamin; jika jarang, jenis kelamin mana yang lebih sering mengidapnya; 2) orang yang berjenis kelamin mewarisi sifat tersebut dari ayah dan ibu yang membawa sifat tersebut.
 Tahapan pemecahan masalah 3. Berdasarkan hasil analisis, cobalah menentukan genotipe seluruh anggota silsilah. Untuk menentukan genotipe, pertama-tama cari tahu rumus pemisahan keturunan dalam satu generasi.
Tahapan pemecahan masalah 3. Berdasarkan hasil analisis, cobalah menentukan genotipe seluruh anggota silsilah. Untuk menentukan genotipe, pertama-tama cari tahu rumus pemisahan keturunan dalam satu generasi.
 Jenis pewarisan suatu sifat. 1. Warisan autosomal dominan: 1) sifat tersebut sering muncul dalam silsilah, di hampir semua generasi, sama seringnya pada anak laki-laki dan perempuan; 2) jika salah satu orang tuanya adalah pembawa suatu sifat, maka sifat tersebut akan muncul pada semua keturunannya atau pada separuhnya.
Jenis pewarisan suatu sifat. 1. Warisan autosomal dominan: 1) sifat tersebut sering muncul dalam silsilah, di hampir semua generasi, sama seringnya pada anak laki-laki dan perempuan; 2) jika salah satu orang tuanya adalah pembawa suatu sifat, maka sifat tersebut akan muncul pada semua keturunannya atau pada separuhnya.
 Glaukoma adalah penyakit mata yang ditandai dengan peningkatan tekanan intraokular dan penurunan ketajaman penglihatan. Faktor risiko berkembangnya glaukoma adalah: keturunan, diabetes melitus, aterosklerosis, trauma mata, penyakit inflamasi dan degeneratif mata. Dengan tekanan intraokular yang terus meningkat, atrofi saraf optik secara bertahap berkembang, dan orang tersebut kehilangan penglihatan. Brachydactyly (brachydactylia; brachy- + jari daktylos Yunani; sinonim jari pendek) adalah anomali perkembangan: pemendekan jari tangan atau kaki. diwariskan secara autosomal dominan.
Glaukoma adalah penyakit mata yang ditandai dengan peningkatan tekanan intraokular dan penurunan ketajaman penglihatan. Faktor risiko berkembangnya glaukoma adalah: keturunan, diabetes melitus, aterosklerosis, trauma mata, penyakit inflamasi dan degeneratif mata. Dengan tekanan intraokular yang terus meningkat, atrofi saraf optik secara bertahap berkembang, dan orang tersebut kehilangan penglihatan. Brachydactyly (brachydactylia; brachy- + jari daktylos Yunani; sinonim jari pendek) adalah anomali perkembangan: pemendekan jari tangan atau kaki. diwariskan secara autosomal dominan.
 Jenis pewarisan suatu sifat. 2. Warisan resesif autosomal: 1) sifat ini jarang terjadi, tidak terjadi pada semua generasi, sama-sama umum pada anak laki-laki dan perempuan; 2) sifat tersebut dapat muncul pada anak, meskipun orang tuanya tidak memiliki sifat tersebut; 3) jika salah satu orang tua merupakan pembawa sifat tersebut, maka sifat tersebut tidak akan muncul pada anak atau akan muncul pada separuh keturunannya.
Jenis pewarisan suatu sifat. 2. Warisan resesif autosomal: 1) sifat ini jarang terjadi, tidak terjadi pada semua generasi, sama-sama umum pada anak laki-laki dan perempuan; 2) sifat tersebut dapat muncul pada anak, meskipun orang tuanya tidak memiliki sifat tersebut; 3) jika salah satu orang tua merupakan pembawa sifat tersebut, maka sifat tersebut tidak akan muncul pada anak atau akan muncul pada separuh keturunannya.
 Apa itu fenilketonuria? Fenilketonuria (PKU) adalah kelainan bawaan yang meningkatkan jumlah asam amino fenilalanin dalam darah ke tingkat yang berbahaya. (Asam amino adalah bahan penyusun protein). Jika PKU tidak diobati, kelebihan fenilalanin dapat menyebabkan keterbelakangan mental dan masalah kesehatan serius lainnya. Bagaimana cara orang mewarisi PKU? PKU diturunkan secara autosomal resesif, yang berarti dua salinan gen harus diubah agar seseorang dapat terkena penyakit tersebut. Seringkali, orang tua dari anak dengan kelainan resesif autosomal tidak terpengaruh, namun merupakan pembawa satu salinan gen yang diubah.
Apa itu fenilketonuria? Fenilketonuria (PKU) adalah kelainan bawaan yang meningkatkan jumlah asam amino fenilalanin dalam darah ke tingkat yang berbahaya. (Asam amino adalah bahan penyusun protein). Jika PKU tidak diobati, kelebihan fenilalanin dapat menyebabkan keterbelakangan mental dan masalah kesehatan serius lainnya. Bagaimana cara orang mewarisi PKU? PKU diturunkan secara autosomal resesif, yang berarti dua salinan gen harus diubah agar seseorang dapat terkena penyakit tersebut. Seringkali, orang tua dari anak dengan kelainan resesif autosomal tidak terpengaruh, namun merupakan pembawa satu salinan gen yang diubah.
 Jenis pewarisan suatu sifat. 3. Pewarisan tertaut jenis kelamin: 1) X - pewarisan dominan: ü sifat tersebut lebih banyak terdapat pada perempuan; ü jika ibu sakit dan bapak sehat, maka sifat tersebut diturunkan kepada keturunannya tanpa memandang jenis kelamin; dapat terwujud baik pada anak perempuan maupun laki-laki; ü Jika ibu sehat dan ayah sakit, maka semua anak perempuan akan menunjukkan gejala tersebut, tetapi anak laki-laki tidak.
Jenis pewarisan suatu sifat. 3. Pewarisan tertaut jenis kelamin: 1) X - pewarisan dominan: ü sifat tersebut lebih banyak terdapat pada perempuan; ü jika ibu sakit dan bapak sehat, maka sifat tersebut diturunkan kepada keturunannya tanpa memandang jenis kelamin; dapat terwujud baik pada anak perempuan maupun laki-laki; ü Jika ibu sehat dan ayah sakit, maka semua anak perempuan akan menunjukkan gejala tersebut, tetapi anak laki-laki tidak.
 3. Warisan terpaut seks: 2) X - pewarisan resesif: sifat tersebut lebih sering ditemukan pada laki-laki; Lebih sering gejalanya muncul setelah satu generasi; Jika kedua orang tuanya sehat, tetapi ibunya heterozigot, maka sifat tersebut sering muncul pada 50% anak laki-laki; Jika ayah sakit dan ibu heterozigot, maka perempuan juga dapat memiliki sifat tersebut.
3. Warisan terpaut seks: 2) X - pewarisan resesif: sifat tersebut lebih sering ditemukan pada laki-laki; Lebih sering gejalanya muncul setelah satu generasi; Jika kedua orang tuanya sehat, tetapi ibunya heterozigot, maka sifat tersebut sering muncul pada 50% anak laki-laki; Jika ayah sakit dan ibu heterozigot, maka perempuan juga dapat memiliki sifat tersebut.

 3. Warisan terkait seks: 3) Warisan terkait Y: üsifat hanya terdapat pada laki-laki; Jika sang ayah membawa suatu sifat, maka, pada umumnya, semua anak laki-laki juga memiliki sifat tersebut.
3. Warisan terkait seks: 3) Warisan terkait Y: üsifat hanya terdapat pada laki-laki; Jika sang ayah membawa suatu sifat, maka, pada umumnya, semua anak laki-laki juga memiliki sifat tersebut.
 Contoh penyelesaian masalah Proband adalah wanita yang tidak kidal. Kedua saudara perempuannya tidak kidal, dan kedua saudara laki-lakinya kidal. Ibu tidak kidal. Dia memiliki dua saudara laki-laki dan satu saudara perempuan, semuanya tidak kidal. Nenek dan kakek tidak kidal. Ayah proband kidal, saudara perempuan dan laki-lakinya kidal, dua saudara laki-laki dan perempuan lainnya kidal. Penyelesaian: 1. Gambarkan simbol probandnya. Kami menunjukkan keberadaan tanda di proband.
Contoh penyelesaian masalah Proband adalah wanita yang tidak kidal. Kedua saudara perempuannya tidak kidal, dan kedua saudara laki-lakinya kidal. Ibu tidak kidal. Dia memiliki dua saudara laki-laki dan satu saudara perempuan, semuanya tidak kidal. Nenek dan kakek tidak kidal. Ayah proband kidal, saudara perempuan dan laki-lakinya kidal, dua saudara laki-laki dan perempuan lainnya kidal. Penyelesaian: 1. Gambarkan simbol probandnya. Kami menunjukkan keberadaan tanda di proband.
 2. Lambang saudara kandungnya kita tempatkan di sebelah lambang proband. Kami menghubungkannya dengan rocker grafis.
2. Lambang saudara kandungnya kita tempatkan di sebelah lambang proband. Kami menghubungkannya dengan rocker grafis.




 7. Menentukan genotipe anggota silsilahnya. Tanda kidal muncul di setiap generasi baik pada wanita maupun pria. Hal ini menunjukkan jenis pewarisan sifat autosomal dominan. I A- A- II A- A- A- Aa aa A- III aa Aa Aa A- aa
7. Menentukan genotipe anggota silsilahnya. Tanda kidal muncul di setiap generasi baik pada wanita maupun pria. Hal ini menunjukkan jenis pewarisan sifat autosomal dominan. I A- A- II A- A- A- Aa aa A- III aa Aa Aa A- aa
 Tugas 2. Berdasarkan silsilah yang ditunjukkan pada gambar, tentukan sifat manifestasi sifat yang ditandai dengan warna hitam (dominan, resesif, terpaut jenis kelamin atau tidak). Tentukan genotipe orang tua dan anak pada generasi pertama.
Tugas 2. Berdasarkan silsilah yang ditunjukkan pada gambar, tentukan sifat manifestasi sifat yang ditandai dengan warna hitam (dominan, resesif, terpaut jenis kelamin atau tidak). Tentukan genotipe orang tua dan anak pada generasi pertama.
 Skema pemecahan masalah: 1) Sifat resesif tidak terpaut seks; 2) Genotipe orang tua: ibu - aa, ayah - AA atau Aa 3) Genotipe anak: putra dan putri heterozigot - Aa.
Skema pemecahan masalah: 1) Sifat resesif tidak terpaut seks; 2) Genotipe orang tua: ibu - aa, ayah - AA atau Aa 3) Genotipe anak: putra dan putri heterozigot - Aa.
 Tugas 3 Dengan menggunakan silsilah yang ditunjukkan pada diagram, tentukan jenis dan sifat manifestasi sifat yang disorot dalam warna hitam (dominan, resesif, terpaut jenis kelamin atau tidak). Tentukan genotipe anak pada generasi pertama.
Tugas 3 Dengan menggunakan silsilah yang ditunjukkan pada diagram, tentukan jenis dan sifat manifestasi sifat yang disorot dalam warna hitam (dominan, resesif, terpaut jenis kelamin atau tidak). Tentukan genotipe anak pada generasi pertama.
 Skema penyelesaian masalah: 1) Sifatnya resesif, terikat pada kromosom X; 2) Genotipe orang tua: ibu – XHA, ayah – XAU; 3) Genotipe anak pada F 1 : anak – Ha. Uh, putri - HAHA putri - HAHA
Skema penyelesaian masalah: 1) Sifatnya resesif, terikat pada kromosom X; 2) Genotipe orang tua: ibu – XHA, ayah – XAU; 3) Genotipe anak pada F 1 : anak – Ha. Uh, putri - HAHA putri - HAHA
 Tugas 4 Dengan menggunakan silsilah seseorang yang ditunjukkan pada gambar, tentukan sifat pewarisan sifat “mata kecil” yang disorot dalam warna hitam (dominan atau resesif, terpaut jenis kelamin atau tidak). Tentukan genotipe induk dan keturunan F 1 (1, 2, 3, 4, 5). 1 2 3 4 5
Tugas 4 Dengan menggunakan silsilah seseorang yang ditunjukkan pada gambar, tentukan sifat pewarisan sifat “mata kecil” yang disorot dalam warna hitam (dominan atau resesif, terpaut jenis kelamin atau tidak). Tentukan genotipe induk dan keturunan F 1 (1, 2, 3, 4, 5). 1 2 3 4 5
 Skema pemecahan masalah: 1) Sifatnya resesif, tidak terpaut seks; 2) Genotipe orang tua: ibu – Aa, ayah – Aa; 3) Genotipe keturunan pada F 1: 1, 2 – Aa, 3, 5 – AA atau Aa; 4 – aa.
Skema pemecahan masalah: 1) Sifatnya resesif, tidak terpaut seks; 2) Genotipe orang tua: ibu – Aa, ayah – Aa; 3) Genotipe keturunan pada F 1: 1, 2 – Aa, 3, 5 – AA atau Aa; 4 – aa.
 Pengkode unsur isi dalam biologi 3.4 Genetika, tugasnya. Keturunan dan variabilitas adalah sifat organisme. Metode genetika. Konsep dasar genetik dan simbolisme. Teori hereditas kromosom. Ide-ide modern tentang gen dan genom. 3. 5 Pola hereditas, dasar sitologinya. Pola pewarisan yang ditetapkan oleh G. Mendel, dasar sitologinya (persilangan mono dan dihibrid). Hukum Morgan: pewarisan sifat yang saling terkait, gangguan hubungan gen. Genetika seks. Warisan sifat-sifat terkait seks. Interaksi gen. Genotipe sebagai suatu sistem integral. Genetika manusia. Metode untuk mempelajari genetika manusia. Memecahkan masalah genetik. Menyusun skema penyeberangan.
Pengkode unsur isi dalam biologi 3.4 Genetika, tugasnya. Keturunan dan variabilitas adalah sifat organisme. Metode genetika. Konsep dasar genetik dan simbolisme. Teori hereditas kromosom. Ide-ide modern tentang gen dan genom. 3. 5 Pola hereditas, dasar sitologinya. Pola pewarisan yang ditetapkan oleh G. Mendel, dasar sitologinya (persilangan mono dan dihibrid). Hukum Morgan: pewarisan sifat yang saling terkait, gangguan hubungan gen. Genetika seks. Warisan sifat-sifat terkait seks. Interaksi gen. Genotipe sebagai suatu sistem integral. Genetika manusia. Metode untuk mempelajari genetika manusia. Memecahkan masalah genetik. Menyusun skema penyeberangan.
 SPESIFIKASI Makalah Ujian Biologi A 7. Genetika, Tugasnya, Konsep Dasar Genetika. A 8. Pola keturunan. Genetika manusia. A 9. Pola variabilitas. A 30. Pola genetik. Pengaruh mutagen pada peralatan genetik sel dan organisme. C 6. Memecahkan masalah genetika untuk menerapkan pengetahuan dalam situasi baru.
SPESIFIKASI Makalah Ujian Biologi A 7. Genetika, Tugasnya, Konsep Dasar Genetika. A 8. Pola keturunan. Genetika manusia. A 9. Pola variabilitas. A 30. Pola genetik. Pengaruh mutagen pada peralatan genetik sel dan organisme. C 6. Memecahkan masalah genetika untuk menerapkan pengetahuan dalam situasi baru.
 Bagian A 1. Genetika sangat penting dalam dunia kedokteran, karena 1) memerangi epidemi 2) menciptakan obat untuk mengobati pasien 3) mengetahui penyebab penyakit keturunan 4) melindungi lingkungan dari pencemaran oleh mutagen
Bagian A 1. Genetika sangat penting dalam dunia kedokteran, karena 1) memerangi epidemi 2) menciptakan obat untuk mengobati pasien 3) mengetahui penyebab penyakit keturunan 4) melindungi lingkungan dari pencemaran oleh mutagen
 2. Metode yang digunakan untuk mempelajari sifat-sifat perwujudan sifat-sifat pada saudara perempuan atau laki-laki yang berkembang dari satu sel telur yang telah dibuahi disebut 1. 2. 3. 4. Kembar Sitogenetik Silsilah Hibridologi
2. Metode yang digunakan untuk mempelajari sifat-sifat perwujudan sifat-sifat pada saudara perempuan atau laki-laki yang berkembang dari satu sel telur yang telah dibuahi disebut 1. 2. 3. 4. Kembar Sitogenetik Silsilah Hibridologi
 3. Metode genealogi digunakan untuk 1) Memperoleh mutasi gen dan genom 2) Mempelajari pengaruh pendidikan terhadap entogenesis manusia 3) Meneliti penyakit keturunan pada manusia 4) Mempelajari tahapan evolusi dunia organik
3. Metode genealogi digunakan untuk 1) Memperoleh mutasi gen dan genom 2) Mempelajari pengaruh pendidikan terhadap entogenesis manusia 3) Meneliti penyakit keturunan pada manusia 4) Mempelajari tahapan evolusi dunia organik
 4. Apa fungsi konsultasi medis genetik pada pasangan orang tua? 1. Mengidentifikasi kecenderungan orang tua terhadap penyakit menular 2. Menentukan kemungkinan mempunyai anak kembar 3. Menentukan kemungkinan terjadinya penyakit keturunan pada anak 4. Mengidentifikasi kecenderungan orang tua terhadap gangguan metabolisme
4. Apa fungsi konsultasi medis genetik pada pasangan orang tua? 1. Mengidentifikasi kecenderungan orang tua terhadap penyakit menular 2. Menentukan kemungkinan mempunyai anak kembar 3. Menentukan kemungkinan terjadinya penyakit keturunan pada anak 4. Mengidentifikasi kecenderungan orang tua terhadap gangguan metabolisme
 Menentukan genotipe berdasarkan fenotipe Warna mata seseorang ditentukan oleh gen autosom; buta warna adalah gen resesif yang terkait dengan jenis kelamin. Tentukan genotipe wanita bermata coklat dengan penglihatan warna normal, ayahnya buta warna (mata coklat mendominasi mata biru) 1) AAXDXD 3) Aa. Xd 2) Aa. XDXd 4) aa. XDXd
Menentukan genotipe berdasarkan fenotipe Warna mata seseorang ditentukan oleh gen autosom; buta warna adalah gen resesif yang terkait dengan jenis kelamin. Tentukan genotipe wanita bermata coklat dengan penglihatan warna normal, ayahnya buta warna (mata coklat mendominasi mata biru) 1) AAXDXD 3) Aa. Xd 2) Aa. XDXd 4) aa. XDXd
 Bagian C Penyelesaian masalah genetik pada penerapan ilmu dalam situasi baru: persilangan dihibrid, pewarisan sifat terpaut jenis kelamin, pewarisan sifat terpaut (dengan pindah silang, tanpa pindah silang), penentuan golongan darah, analisis silsilah
Bagian C Penyelesaian masalah genetik pada penerapan ilmu dalam situasi baru: persilangan dihibrid, pewarisan sifat terpaut jenis kelamin, pewarisan sifat terpaut (dengan pindah silang, tanpa pindah silang), penentuan golongan darah, analisis silsilah
 Bagian C Pada manusia, pewarisan albinisme tidak terkait seks (A - adanya melanin dalam sel kulit, dan - tidak adanya melanin dalam sel kulit - albinisme), dan hemofilia terkait seks (XH - pembekuan darah normal , Xh - hemofilia). Tentukan genotipe orang tua, serta kemungkinan genotipe, jenis kelamin dan fenotipe anak dari perkawinan wanita dihomozigot, normal kedua alelnya, dan pria albino penderita hemofilia. Buatlah diagram untuk menyelesaikan masalah tersebut.
Bagian C Pada manusia, pewarisan albinisme tidak terkait seks (A - adanya melanin dalam sel kulit, dan - tidak adanya melanin dalam sel kulit - albinisme), dan hemofilia terkait seks (XH - pembekuan darah normal , Xh - hemofilia). Tentukan genotipe orang tua, serta kemungkinan genotipe, jenis kelamin dan fenotipe anak dari perkawinan wanita dihomozigot, normal kedua alelnya, dan pria albino penderita hemofilia. Buatlah diagram untuk menyelesaikan masalah tersebut.
 Skema penyelesaian masalah meliputi: 1) genotipe tetua: ♀AAXHXH (gamet AXH); ♂aa. Xh. Y (gamet a.Xh, a.Y); 2) genotipe dan jenis kelamin anak: ♀Aa. XHXh; ♂Aa. XHY; 3) fenotipe anak-anak: seorang gadis yang secara lahiriah normal untuk kedua alelnya, tetapi merupakan pembawa gen albinisme dan hemofilia; Seorang anak laki-laki yang secara lahiriah normal untuk kedua alelnya, tetapi merupakan pembawa gen albinisme.
Skema penyelesaian masalah meliputi: 1) genotipe tetua: ♀AAXHXH (gamet AXH); ♂aa. Xh. Y (gamet a.Xh, a.Y); 2) genotipe dan jenis kelamin anak: ♀Aa. XHXh; ♂Aa. XHY; 3) fenotipe anak-anak: seorang gadis yang secara lahiriah normal untuk kedua alelnya, tetapi merupakan pembawa gen albinisme dan hemofilia; Seorang anak laki-laki yang secara lahiriah normal untuk kedua alelnya, tetapi merupakan pembawa gen albinisme.
Anomali yang mengarah ke peningkatan level fenilalanin darah, paling sering defisiensi fenilalanin hidroksilase (PAH) atau fenilketonuria (PKU), menggambarkan hampir semua prinsip genetika biokimia yang berkaitan dengan kerusakan enzim. Semua kelainan genetik pada metabolisme fenilalanin adalah akibat hilangnya mutasi fungsi pada gen yang mengkode PAH atau pada gen yang diperlukan untuk sintesis atau pemulihan kofaktornya, BH4.
Fenilketonuria klasik(PKU) dianggap sebagai contoh teladan kesalahan metabolisme bawaan. Ini adalah kelainan pemecahan fenilalanin resesif autosomal yang disebabkan oleh mutasi pada gen yang mengkode PAH, enzim yang mengubah fenilalanin menjadi tirosin. Penemuan fenilketonuria (PKU) oleh Fehling pada tahun 1934 merupakan penemuan pertama yang menunjukkan adanya cacat genetik sebagai penyebab keterbelakangan mental.
Karena ketidakmampuan untuk mendaur ulang fenilalanin pasien dengan fenilketonuria (PKU) menumpuk asam amino ini dalam cairan tubuh. Hiperfenilalaninemia merusak sistem saraf pusat yang sedang berkembang pada anak usia dini dan mengganggu fungsi otak dewasa. Sebagian kecil fenilalanin dimetabolisme melalui jalur alternatif, menghasilkan peningkatan jumlah asam fenilpiruvat (asam keto yang menjadi nama penyakit ini) dan metabolit lain yang diekskresikan dalam urin.
Namun menariknya cacat enzim telah diketahui selama beberapa dekade, mekanisme patogenetik yang tepat mengenai bagaimana peningkatan fenilalanin merusak otak masih belum diketahui. Yang penting, perkembangan kerusakan neurologis yang disebabkan oleh blok metabolik pada PKU klasik sebagian besar dapat dicegah dengan perubahan pola makan yang mencegah akumulasi fenilalanin. Pengobatan fenilketonuria (PKU) telah menjadi model pengobatan banyak penyakit metabolik, yang hasilnya dapat ditingkatkan dengan mencegah akumulasi substrat enzim dan turunannya.
Skrining bayi baru lahir untuk fenilketonuria (PKU)
Populasi banyak digunakan penyaringan bayi baru lahir karena fenilketonuria (PKU). Fenilketonuria (PKU) adalah contoh penyakit genetik yang memerlukan skrining massal pada neonatus; penyakit ini relatif umum terjadi pada sejumlah populasi (hingga 1 dari 2.900 bayi baru lahir hidup). Perawatan yang dimulai sejak dini sangat efektif; tanpa pengobatan, keterbelakangan mental yang parah pasti akan berkembang. Tes skrining dilakukan beberapa hari setelah kelahiran.
Setetes darah diperoleh dari tusukan tumit, diaplikasikan pada kertas saring, dikeringkan dan dikirim ke laboratorium terpusat untuk menilai kadar fenilalanin darah dan rasio fenilalanin/tirosin. Dulu, sampel dikumpulkan sebelum bayi keluar dari rumah sakit. Kecenderungan pulangnya ibu dan bayi baru lahir lebih awal setelah melahirkan telah mengubah praktik ini. Sebaiknya tidak melakukan tes sebelum usia 24 jam karena kadar fenilalanin pada fenilketonuria (PKU) tidak meningkat sampai setelah lahir. Hasil tes yang positif harus segera dipastikan, karena menunda memulai pengobatan lebih dari 4 minggu setelah melahirkan tidak menghindari dampak pada status intelektual pasien dengan fenilketonuria (PKU).
Berbagai bentuk fenilketonuria dan hiperfenilalaninemia
Karena (PKU) dikaitkan dengan defisiensi aktivitas fenilalanin hidroksilase (PAH) yang parah (kurang dari 1% dibandingkan dengan kontrol), PAH mutan dengan aktivitas sisa menyebabkan manifestasi fenotipik yang tidak terlalu parah, yang disebut hiperfenilalaninemia dan fenilketonuria atipikal (PKU).
Hiperfenilalaninemia fenilketonuria (PKU), selain fenilketonuria (PKU), didiagnosis jika konsentrasi fenilalanin plasma di bawah 1 mmol/L dengan adanya pola makan normal. Derajat hiperfenilalaninemia ini hanya 10 kali lebih tinggi dari normal dan secara signifikan lebih rendah dibandingkan konsentrasi yang ditemukan pada fenilketonuria klasik (PKU) (>1 mmol/L). Peningkatan moderat fenilalanin pada hiperfenilalaninemia kemungkinan besar tidak membahayakan fungsi otak dan bahkan mungkin bermanfaat jika peningkatannya kecil (<0,4 ммоль), такие дети обращают на себя внимание врачей только благодаря скринингу. Их нормальный фенотип оказался наилучшим показателем безопасного уровня фенилаланина плазмы, который не следует превышать при лечении пациентов с классической фенилкетонурии (ФКУ).
Tidak lazim(PKU) - kategori yang mencakup pasien dengan kadar fenilalanin antara PKU klasik dan hiperfenilalaninemia; pasien seperti itu memerlukan pembatasan fenilalanin dalam makanannya, tetapi lebih sedikit dibandingkan pasien dengan fenilketonuria klasik (PKU). Kompleks ketiga fenotip klinis dengan mutasi pada gen PAH merupakan contoh heterogenitas klinis.
Hiperfenilalaninemia: heterogenitas alelik dan lokus pada fenilketonuria (PKU)
Molekuler cacat pada gen fenilalanin hidroksilase. Pasien dengan hiperfenilalaninemia, termasuk fenilketonuria klasik (PKU), fenilketonuria atipikal (PKU), dan hiperfenilalaninemia jinak, menunjukkan tingkat heterogenitas alelik yang mencolok pada lokus fenilalanin hidroksilase (PAH) (lebih dari 400 mutasi berbeda di seluruh dunia).
Sebagian besar alel fenilalanin hidroksilase(PAH) adalah mutasi yang cukup langka yang mengganggu sifat enzimatik fenilalanin hidroksilase (PAH) dan menyebabkan hiperfenilalaninemia, meskipun polimorfisme jinak atau varian jinak yang kurang umum juga telah ditemukan.
Dalam populasi Keturunan Eropa sekitar dua pertiga dari kromosom mutan yang diketahui diwakili oleh enam mutasi. Enam mutasi lainnya bertanggung jawab atas lebih dari 80% mutasi fenilalanin hidroksilase (PAH) pada populasi Asia. Mutasi patogen lainnya lebih jarang terjadi. Agar informasi ini tersedia secara luas, sebuah konsorsium internasional telah mengembangkan database mutasi pada gen fenilalanin hidroksilase (PAH).
Secara keseluruhan populasi Ada heterogenitas genetik yang nyata dari fenilalanin hidroksilase (PAH). Karena tingginya tingkat heterogenitas alel di lokus, sebagian besar pasien dengan fenilketonuria (PKU) di banyak populasi adalah heterozigot majemuk (yaitu, mereka memiliki dua alel patogen yang berbeda), yang sepenuhnya konsisten dengan heterogenitas enzimatik dan fenotipik yang diamati di kelainan fenilalanin hidroksilase (PAH).

Pada awalnya tampak seperti pengetahuan tentang genotipe fenilalanin hidroksilase(FA) secara andal memprediksi detail fenotipe; Harapan ini tidak sepenuhnya dapat dibenarkan, meskipun ditemukan korelasi tertentu antara genotipe PAH dan fenotipe biokimia.
Secara umum, mutasi yang sepenuhnya menekan atau mengurangi aktivitas secara drastis fenilalanin hidroksilase(PAH) menyebabkan fenilketonuria klasik (PKU), sedangkan mutasi yang mengakibatkan aktivitas enzim sisa yang cukup besar berhubungan dengan fenotip ringan.
Namun, beberapa mutasi fenilalanin hidroksilase(FA) pada pasien homozigot menentukan seluruh spektrum fenotip, dari fenilketonuria klasik (PKU) hingga hiperfenilalaninemia jinak.
Dengan demikian, menjadi jelas bahwa dalam formasi fenotip diamati pada genotipe tertentu, faktor biologis lain yang tidak teridentifikasi terlibat, tidak diragukan lagi termasuk gen pengubah. Pengamatan ini, yang kini diakui sebagai karakteristik umum dari banyak penyakit monogenik, menunjukkan bahwa penyakit monogenik seperti fenilketonuria (PKU) bukanlah penyakit yang secara genetik sederhana.
Cacat metabolisme tetrahidrobiopterin pada fenilketonuria (PKU)
Awalnya diyakini bahwa semua anak adalah keturunan hiperfenilalaninemia mengalami defisiensi fenilalanin hidroksilase (PAH) primer. Sekarang jelas bahwa sekitar 1-3% pasien memiliki gen PAH yang normal dan hiperfenilalaninemianya merupakan akibat dari cacat genetik pada salah satu dari beberapa gen lain yang terlibat dalam sintesis atau regenerasi kofaktor PAH, BH4. Asosiasi satu fenotipe, seperti hiperfenilalaninemia, dengan mutasi pada gen berbeda merupakan contoh heterogenitas lokus.
Seperti yang ditunjukkan oleh mutasi pada gen penyandi protein fenilalanin hidroksilase(PAH) dan metabolisme kofaktor biopterinnya, protein yang dikodekan oleh gen yang menunjukkan heterogenitas lokus, biasanya berpartisipasi dalam rantai reaksi biokimia yang sama. Pasien dengan defisiensi BH4 pertama kali diidentifikasi karena, meskipun berhasil mempertahankan konsentrasi fenilalanin yang rendah dalam makanannya, mereka mengalami masalah neurologis yang parah sejak dini.
Hasil buruk ini sebagian dapat dijelaskan kebutuhan kofaktor BH4 untuk aktivitas dua enzim lainnya, tirosin hidroksilase dan triptofan hidroksilase. Kedua hidroksilase ini sangat penting untuk sintesis neurotransmiter monoamina seperti dehidroksifenilalanin, norepinefrin, epinefrin, dan serotonin. Pasien dengan defisiensi BH4 mengalami gangguan biosintesis dari GTP atau regenerasi BH4. Seperti fenilketonuria klasik (PKU), kelainan ini diturunkan secara resesif autosomal.
Penting untuk membedakan pasien dengan kelainan metabolisme BH4 dari pasien dengan mutasi fenilalanin hidroksilase(FA), karena perlakuan mereka sangat berbeda. Pertama, karena struktur protein fenilalanin hidroksilase (PAH) normal pada pasien dengan kelainan BH4, aktivitasnya dapat dipulihkan jika pasien tersebut diberi BH4 dosis besar, yang menyebabkan penurunan kadar fenilalanin plasma. Oleh karena itu, tingkat pembatasan fenilalanin dalam makanan pasien dengan kelainan metabolisme BH4 dapat dikurangi secara signifikan, dan beberapa pasien dapat dialihkan ke diet normal (yaitu, tanpa pembatasan fenilalanin).
Kedua, Anda juga harus mencoba normalisasi tingkat neurotransmitter di otak pasien ini dengan pemberian produk tirosin hidroksilase dan triptofan hidroksilase: masing-masing L-dopa dan 5-hydroxytryptophan. Untuk alasan ini, semua bayi baru lahir dengan hiperfenilalaninemia harus dievaluasi untuk kelainan metabolisme BH4.
Reaksi terhadap tetrahydrobiopterin dengan mutasi pada gen PAH pada fenilketonuria (PKU)
Pada sebagian besar pasien dengan mutasi pada gen fenilalanin hidroksilase(PAH), dan bukan dalam metabolisme BH4, terdapat penurunan yang jelas pada kadar fenilalanin dalam darah selama pemberian oral kofaktor fenilalanin hidroksilase (PAH) BH4 dosis besar. Pasien dengan aktivitas sisa fenilalanin hidroksilase (PAH) yang signifikan (yaitu, pasien dengan fenilketonuria atipikal (PKU) dan hiperfenilalaninemia) memberikan respons terbaik terhadap pengobatan tersebut, namun sejumlah kecil pasien bahkan dengan fenilketonuria klasik (PKU) juga merespons pengobatan ini. Pada saat yang sama, adanya aktivitas sisa PAH tidak menjamin efek pada kadar fenilalanin plasma ketika BH4 diresepkan.
Kemungkinan besar tingkat responsnya reaksi pada BH4 tergantung pada sifat spesifik dari setiap protein mutan fenilalanin hidroksilase (PAH), yang mencerminkan heterogenitas alelik yang mendasari mutasi PAH. Telah terbukti bahwa pengenalan BH4 ke dalam makanan memiliki efek terapeutik melalui beberapa mekanisme yang disebabkan oleh peningkatan jumlah kofaktor normal yang bersentuhan dengan kofaktor mutan.
Mekanisme ini termasuk stabilisasi mutan enzim, perlindungan enzim dari degradasi sel, peningkatan pasokan kofaktor ke enzim yang memiliki afinitas rendah terhadap BH4, dan efek menguntungkan lainnya pada sifat kinetik dan katalitik enzim. Memberikan peningkatan jumlah kofaktor adalah strategi umum yang digunakan dalam pengobatan banyak kesalahan metabolisme bawaan.
Sejumlah mutasi gen, yang mengubah struktur hanya satu gen, menyebabkan perkembangan keterbelakangan mental. Menurut beberapa perkiraan, pada 7-10% pasien oligofrenia disebabkan oleh mutasi semacam ini.
Totalitas reaksi biokimia yang terjadi dalam tubuh disebut metabolisme. Banyak gen mengkode protein yang berpartisipasi sebagai enzim dalam reaksi metabolisme tertentu. Mutasi pada gen tersebut dapat menyebabkan tubuh memproduksi enzim yang kurang aktif atau tidak aktif sama sekali, dan terkadang terhentinya sintesis enzim sepenuhnya. Dalam hal ini, reaksi yang biasanya dilakukan oleh enzim ini melambat atau tidak terjadi sama sekali, yang menyebabkan kelainan bawaan yang sesuai - salah satu yang disebut kesalahan metabolisme bawaan. Penyakit keturunan genetik yang paling umum termasuk fenilketonuria, anemia sel sabit, penyakit Tay-Sachs, hemofilia, dan diabetes mellitus. Sejauh mana pengaruhnya terhadap fenotipe bergantung pada seberapa penting enzim yang terpengaruh bagi organisme. Kita telah melihat di atas bahwa penyakit Tay-Sachs dan fibrosis kistik menyebabkan kematian. Beberapa kelainan genetik lainnya menyebabkan berbagai masalah serius pada tubuh, namun tidak berakibat fatal.
Fenilketonuria dan albinisme mempengaruhi jalur metabolisme yang sama.
Fenilketonuria adalah penyakit di mana akibat mutasi, struktur enzim yang terlibat dalam metabolisme asam amino fenilalanin (fenilalanin hidroksilase) terganggu. Enzim ini diperlukan untuk konversi fenilalanin menjadi tirosin. Penyakit semacam ini disebut enzymopathies, yaitu. disebabkan oleh kerusakan pada enzim. Dengan penyakit ini, fenilalanin dan produk metabolisme yang tidak tepat (asam fenilasetat) menumpuk di dalam darah, yang menyebabkan kerusakan pada sistem saraf yang sedang berkembang. Hal ini terutama disebabkan oleh penghancuran mielin dan degenerasi sistem saraf spongiformis. Terjadi keterbelakangan mental, mikrosefali, psikosis, tremor, aktivitas kejang, dan spastisitas.
Fenilketonuria mempengaruhi individu yang homozigot karena gen resesif yang membuat mereka kehilangan kemampuan untuk mensintesis salah satu enzim yang diperlukan untuk mengubah asam amino fenilalanin menjadi asam amino lain, tirosin. Alih-alih diubah menjadi tirosin, fenilalanin diubah menjadi asam fenilpiruvat, yang terakumulasi dalam jumlah beracun di dalam darah, mempengaruhi otak dan (jika tidak segera ditangani) menyebabkan keterbelakangan mental. Urin pasien juga mengandung asam fenilpiruvat, yang memberikan bau khas. Saat ini, fenilketonuria diobati dengan diet khusus. Untuk melakukan ini, pada tahun-tahun pertama kehidupan seorang anak, fenilalanin hampir sepenuhnya dikeluarkan dari makanannya. Setelah perkembangan otak selesai, pasien dengan fenilketonuria menjalani diet normal, namun wanita dengan kelainan genetik ini harus mengikuti diet rendah fenilalanin selama kehamilan untuk mencegah perkembangan abnormal otak janin. Di Amerika Serikat, di banyak negara bagian, semua bayi baru lahir diharuskan menjalani tes khusus untuk PKU dan beberapa kesalahan metabolisme bawaan lainnya.
Individu yang homozigot untuk gen albinisme kekurangan enzim yang biasanya mengkatalisis konversi tirosin menjadi melanin, yaitu. pigmen yang menentukan warna coklat atau hitam pada mata, rambut dan kulit. Albino memiliki rambut putih dan kulit serta mata yang sangat terang. Tentu saja, pertanyaan yang mungkin timbul apakah pasien dengan fenilketonuria juga albino, karena tubuh mereka tidak memproduksi tirosin, yang pada akhirnya menghasilkan melanin. Namun, pasien tersebut bukanlah albino, karena tirosin tidak hanya terbentuk di dalam tubuh sendiri dari fenilalanin, tetapi juga masuk ke dalam tubuh melalui makanan. Benar, pasien dengan fenilketonuria biasanya bermata cerah, berkulit putih, dan berambut pirang. Tentu saja, mungkin ada albino di antara mereka, tetapi hanya jika individu tersebut homozigot untuk kedua gen resesif.
Pewarisan fenilketonuria (PKU) menjelaskan hukum pemisahan. Mutasi ini bersifat resesif, yaitu dapat menolak fenotipe hanya dalam keadaan homozigot. Insiden fenilketonuria tertinggi terjadi di Irlandia (16,4 kasus per 100 ribu bayi baru lahir); sebagai perbandingan: di AS - 5 kasus per 100 ribu bayi baru lahir.
Gen PKU dan varian strukturalnya, yang ditemukan pada populasi berbeda, telah dipelajari dengan baik. Pengetahuan yang kami miliki memungkinkan kami melakukan diagnosis pra-kelahiran secara tepat waktu untuk menentukan apakah embrio yang sedang berkembang mewarisi dua salinan alel PKU dari kedua orang tuanya (fakta pewarisan tersebut secara tajam meningkatkan kemungkinan penyakit). Di beberapa negara, misalnya di Italia, yang angka kejadian PKU cukup tinggi, diagnosis seperti itu wajib dilakukan oleh setiap ibu hamil.
PKU lebih sering terjadi pada mereka yang menikah dengan saudara sedarah. Meskipun kejadian PKU relatif rendah, sekitar 1 dari 50 orang adalah pembawa alel PKU. Probabilitas salah satu pembawa alel PKU akan menikah dengan pembawa alel lain yang sama adalah sekitar 2%. Namun, ketika perkawinan terjadi antara saudara sedarah (yaitu, jika pasangan tersebut memiliki silsilah yang sama di mana alel PKU diwariskan), kemungkinan besar kedua pasangan akan menjadi pembawa alel PKU dan secara bersamaan mewariskan dua alel ke masa depan. anak akan menjadi jauh lebih tinggi 2%.
Dalam kasus fenilketonuria, kita memiliki contoh yang mencolok tentang bagaimana perkembangan penyakit yang bersifat genetik dapat dicegah dengan memilih pengaruh lingkungan. Saat ini, fenilketonuria mudah dideteksi melalui pemeriksaan rutin bayi baru lahir pada usia 2-3 hari (biasanya konsentrasi fenilalanin dalam plasma darah tidak boleh melebihi 4 mg/dL). Pasien menjalani diet rendah fenilalanin, yang membantu menghindari kerusakan perkembangan pada sistem saraf. Dalam hal ini, tirosin menjadi asam amino esensial dan perlu dipastikan keberadaannya dalam makanan. Masa paling kritis adalah tahap awal entogenesis, oleh karena itu, di masa dewasa, banyak yang tidak lagi mematuhi pantangan makanan, meskipun hal ini tetap diinginkan. Wanita dengan fenilketonuria, apapun kondisinya, harus mengikuti diet khusus selama kehamilan, jika tidak, tingginya kadar fenilalanin dalam darahnya akan berdampak buruk pada perkembangan janin.
Fenilketonuria adalah contoh yang baik dari interaksi genotipe-lingkungan. Inti dari penyakit ini terletak pada perbedaan kepekaan individu dengan genotipe yang berbeda terhadap pengaruh lingkungan. Lingkungan yang sama (dalam hal ini lingkungan adalah sifat nutrisi) menyebabkan penyakit parah (fenilketonuria) pada beberapa genotipe, sedangkan pada genotipe lain sama sekali tidak ada perubahan patologis yang diamati. Dalam kondisi lingkungan lain (sesuai dengan pola makan khusus), perbedaan antara genotipe untuk sifat ini (fenilketonuria) hilang.